Publications
Superlubric sliding in a multi-contact fullerene-molybdenum disulfide heterostructure
D. Zhou, A.K. Gupta, K. Bi, A.S. de Wijn, P. Schall
Tribology International 214C 111317 (2026).
D. Zhou, A.K. Gupta, K. Bi, A.S. de Wijn, P. Schall
Tribology International 214C 111317 (2026).
Recent research shows that superlubric 2D materials offer great potential for friction reduction, yet their requirements of flatness and small scale hinder real-world applications. Here, we study a multi-contact van der Waals heterostructure, composed of fullerene (C60) molecules sandwiched between molybdenum disulfide (MoS2) atomic layers by molecular-dynamics simulations. We show that despite the multi-contact geometry, the confined layer of C60 nanoparticles exhibits superlubric behavior at high fullerene density or high applied normal force, where the fullerenes’ rotational degrees of freedom are suppressed and the MoS2 layer slides over the fullerenes. At lower fullerene density or load, fullerene rolling leads to increased frictional dissipation. We demonstrate that the atomic stacking at the fullerene-MoS2 contact evolves from aligned into misaligned with increasing fullerene coverage, reflecting the increasing fullerene-fullerene interactions suppressing their rolling degrees of freedom. Thus, counterintuitively, in the fullerene-MoS2 heterojunction system, rolling of the fullerenes is not beneficial for friction reduction, as superlubric slip offers an alternative, lower-friction mechanism. These results on the multi-contact sliding of a heterojunction of fullerene nanoparticles and 2D material are useful for guiding nanoscale superlubric properties into macroscopic scale superlubric applications.
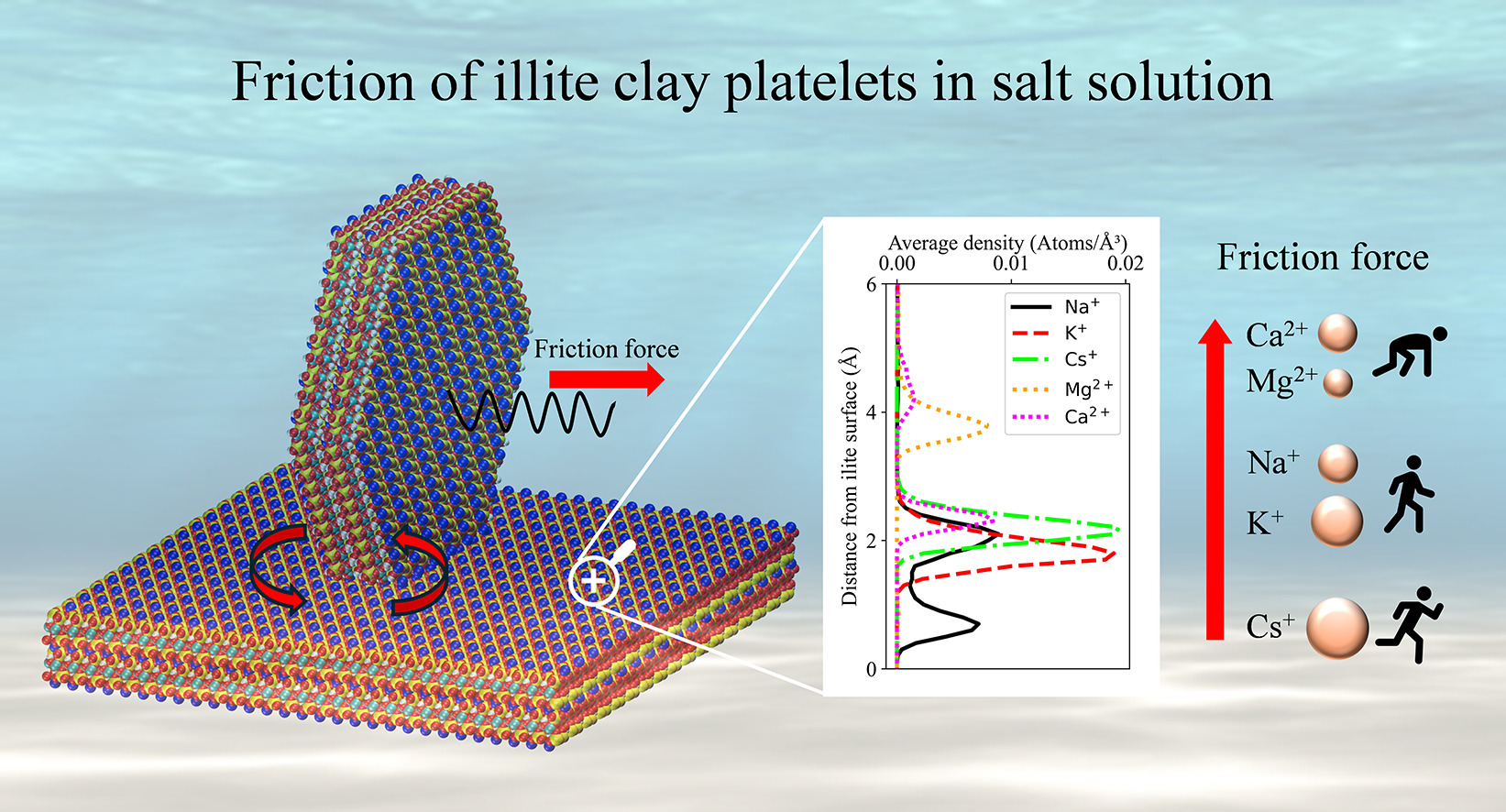
Molecular dynamics simulations of nanoscale friction on illite clay: Effects of solvent salt ions and electric double layer
Ge Li and Astrid S. de Wijn
Journal of Colloid and Interface Science, Volume 703, Part 1, February 2026, 139107.
Ge Li and Astrid S. de Wijn
Journal of Colloid and Interface Science, Volume 703, Part 1, February 2026, 139107.
Quick clay is a highly sensitive soil that transforms rapidly from solid to liquid under minor stress, as a result of long-term salt leaching that drastically reduces shear strength. Stabilizing it is both costly and carbon-intensive, significantly impacting construction emissions in regions like Norway. Developing greener stabilization methods is challenging due to limited understanding of the weakening mechanisms and the specific roles of different salts. In this study, we use molecular dynamics (MD) simulations to investigate the sliding behavior of illite platelets, the key component in Norwegian quick clay, and how it is affected by the different ions in the solution surrounding the surface. We examine the impact of monovalent (NaCl, KCl, CsCl) and divalent (MgCl2 and CaCl2) salts on platelet-surface interactions, focusing on the friction enhancement brought by divalent salts and how the electric double layer (EDL) structure mediates frictional behavior. We find that divalent cations sit higher on top of the surface, and lead to an increase in friction, while monovalent cations sit closer to the surface. By providing a detailed analysis of these interactions, the study offers a novel framework for understanding the role of salts in clay mechanics and highlights opportunities to design environmentally friendly stabilizers as alternatives to traditional lime and cement.
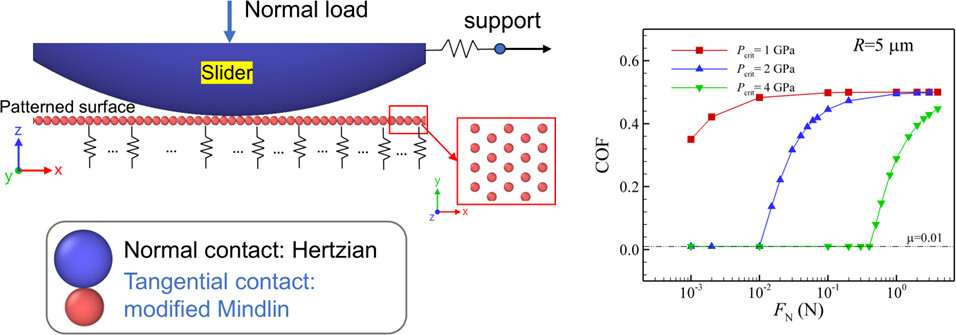
A Theoretical Study on Friction of Macroscale Patterned Surfaces: Implications for Scaling Up Superlubricity
Viet Hung Ho, Melisa M. Gianetti, Ahmed Uluca, Aaron D. Sinnott, Bjørn Haugen, Graham L. W. Cross, and Astrid S. de Wijn
ACS Appl. Mater. Interfaces 2025, 17, 40, 56661–56671.
Viet Hung Ho, Melisa M. Gianetti, Ahmed Uluca, Aaron D. Sinnott, Bjørn Haugen, Graham L. W. Cross, and Astrid S. de Wijn
ACS Appl. Mater. Interfaces 2025, 17, 40, 56661–56671.
“Structural superlubricity”, a state of frictionless sliding between crystalline surfaces, has been observed at the nanoscale and microscale. However, achieving it at the macroscale requires further investigation. Inspired by recent experimental studies, we theoretically examine the friction behavior of macroscale patterned surfaces composed of microscale bumps coated with superlubricious two-dimensional materials. We performed numerical simulations with the discrete element method. The Hertz contact model, along with a modified tangential Mindlin contact model, is employed to capture the nonlinear relationship between the coefficient of friction and normal load. Our results reveal that the friction behavior is significantly influenced by the radius of the microscale bumps, the durability of the coating, and the elasticity of the surface, and we show how those can be tuned to improve friction properties. Additionally, we analytically investigate the deformation mechanisms of the surface structure and derive scaling laws for parameters and the breakdown of superlubricity. The simulation results show strong agreement with the analytical derivations of power laws for scaling of various quantities with the total macroscopic load. Finally, we examine imperfect conditions by investigating how height variations impact frictional performance.

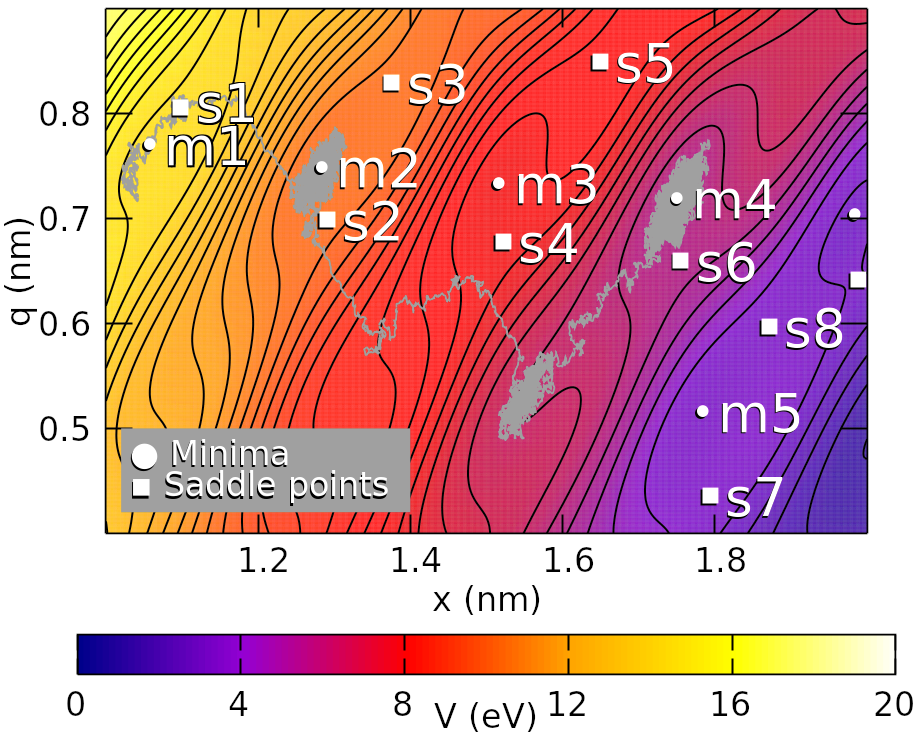
Decay Behaviour, State Transitions and Escape Rates in Simple Models for Complex Systems
Jennifer Sheehan
PhD thesis, Norwegian University of Science and Technology (2025).
Jennifer Sheehan
PhD thesis, Norwegian University of Science and Technology (2025).
Describing phenomena through the lens of decay behaviour, state transitions and escape rates can be applied to many topics across a plethora of disciplines in science and everyday life. In this thesis, I will offer two examples of such cases in physics and biology in different ways. This is done via the construction of simple models for the complex systems that are investigated. In the first topic where this is applied, spontaneous frictional relaxation in atomically thin layered materials is studied. The model for this system was developed by my co-authors, and it mathematically describes the frictional behaviour of atomically thin, layered materials - e.g. graphene. The model mimics an atomic force microscope contact-sliding on a surface. Once this model is subject to temperature and sliding is stopped, the system relaxes via thermal activation. This thermal activation is examined in terms of calculable escape rates and validated using the decay behaviour of the system. The second topic is a molecular model for a keystone mechanism in one type of blood cancer, chronic myeloid leukaemia (CML). The mechanism in focus is the phosphorylation of proteins by Abl1 (an enzyme). The most common treatment for CML is with Abl1 inhibitors or allosteric regulators (specifically imatinib, ponatinib, dasatinib, nilotinib and asciminib in this work). After creating a simple model for this Abl1 enzyme system, the transitions in the system are modelled as decay behaviour. The results of this model for mono-therapy with imatinib, ponatinib and dasatinib lead to the suggestion of an improved method of comparing treatment options for patients experiencing resistance to their current therapy. The model was then extended to include combination therapy of either imatinib, ponatinib, or nilotinib with asciminib, an allosteric regulator. In cases of low synergy or antagonism at patient-relevant treatment concentrations, the improved treatment comparison method for mono-therapy is easily applied for combination therapies.
A computational dynamic model of combination treatment for type II inhibitors with asciminib
J. Roadnight Sheehan, Astrid S. de Wijn, and Ran Friedman
Protein Science, Volume 34, Issue 8 August 2025, e70219 .
J. Roadnight Sheehan, Astrid S. de Wijn, and Ran Friedman
Protein Science, Volume 34, Issue 8 August 2025, e70219 .
Despite continuous strides forward in drug development, resistance to treatment looms large in the battle against cancer as well as communicable diseases. Chronic myeloid leukemia (CML) is treated with targeted therapy and treatment is personalized when resistance arises. It has been extensively studied and is used as a model for targeted therapy. In this study, we examine combination treatments of type II Abl1 inhibitors and asciminib (an allosteric regulator) through a computational model at patient relevant concentrations. Due to the separate binding sites of type II inhibitors and asciminib, we propose their combination treatment as potentially robust to resistance. We find that the simultaneous cobinding of type II inhibitors and asciminib is high in synergetic combinations. As an aid to designing and comparing combination treatments, we put forward an equation that expands on the previously published effective ratio of IC50 (ERIC). Unlike usual comparisons of IC50 values, ERIC takes patient plasma concentrations into account. This study shows that the product of two ERIC values ( ERIC combo ) creates comparable approximations of the effectiveness of combination treatments with low levels of synergy or antagonism at different concentrations. Its simple formulation is done without experiments and requires less computation and input data than the current standard of ZIP values. As such, the new scheme is a useful complement to experiments that deal with synergy in drug use.
Atomic alignment and friction behavior of dense fullerene packings revealed by molecular dynamics simulations
D. Zhou, A. K. Gupta, V. H. Ho, K. Bi, M. M. Gianetti, A. S. de. Wijn, and P. Schall
Friction, 2025.
D. Zhou, A. K. Gupta, V. H. Ho, K. Bi, M. M. Gianetti, A. S. de. Wijn, and P. Schall
Friction, 2025.
Recent experimental and simulation work explores the possibility of scaling up the microscopic phenomenon of superlubricity to the macroscopic scale. Here, we investigate the lubrication behavior of dense packings of C60 fullerenes sandwiched in between two rigid fullerene slabs using atomistic simulations. Using a range of atomic potentials common for carbon-based nanomaterials, we investigate the fullerenes’ atomic stacking and resulting friction properties of the packing as a function of boundary roughness and applied normal load. We find superlubric behavior for flat boundaries due to boundary slip, but finite friction for rough boundaries, due to bulk shear and the related energy dissipation in the bulk. The atomistic simulations reveal a preferred AA-type atomic stacking, which changes to TA-type stacking as the applied load is increased. This is accompanied by a loss of rolling motion of the particles in the highly condensed sheared packing. These results provide atomistic insight into the collective interactions of superlubric particles that exhibit many rotational and translational degrees of freedom in dense packings, and reveal their emergent frictional properties for friction-reduction applications.

Beyond IC50 - A computational dynamic model of drug resistance in enzyme inhibition treatment
J. Roadnight Sheehan, Astrid de Wijn, Thales Souza Freire, Ran Friedman
Plos Computational Biology (2024).
J. Roadnight Sheehan, Astrid de Wijn, Thales Souza Freire, Ran Friedman
Plos Computational Biology (2024).
Resistance to therapy is a major clinical obstacle to treatment of cancer and communicable diseases. Drug selection in treatment of patients where the disease is showing resistance to therapy is often guided by IC50 or fold-IC50 values. In this work, through a model of the treatment of chronic myeloid leukaemia (CML), we contest using fold-IC50 values as a guide for treatment selection. CML is a blood cancer that is treated with Abl1 inhibitors, and is often seen as a model for targeted therapy and drug resistance. Resistance to the first-line treatment occurs in approximately one in four patients. The most common cause of resistance is mutations in the Abl1 enzyme. Different mutant Abl1 enzymes show resistance to different Abl1 inhibitors and the mechanisms that lead to resistance for various mutation and inhibitor combinations are not fully known, making the selection of Abl1 inhibitors for treatment a difficult task. We developed a model based on information of catalysis, inhibition and pharmacokinetics, and applied it to study the effect of three Abl1 inhibitors on mutants of the Abl1 enzyme. From this model, we show that the relative decrease of product formation rate (defined in this work as “inhibitory reduction prowess”) is a better indicator of resistance than an examination of the size of the product formation rate or fold-IC50 values for the mutant. We also examine current ideas and practices that guide treatment choice and suggest a new parameter for selecting treatments that could increase the efficacy and thus have a positive impact on patient outcomes.
Extended kinetic theory applied to pressure-controlled shear flows of frictionless spheres between rigid, bumpy planes
Dalila Vescovi, Astrid S. de Wijn, Graham L. W. Cross, and Diego Berzi
Soft Matter, 2024, 20, 8702-8715.
Dalila Vescovi, Astrid S. de Wijn, Graham L. W. Cross, and Diego Berzi
Soft Matter, 2024, 20, 8702-8715.
We numerically investigate, through discrete element simulations, the steady flow of identical, frictionless spheres sheared between two parallel, bumpy planes in the absence of gravity and under a fixed normal load. We measure the spatial distributions of solid volume fraction, mean velocity, intensity of agitation and stresses, and confirm previous results on the validity of the equation of state and the viscosity predicted by the kinetic theory of inelastic granular gases. We also directly measure the spatial distributions of the diffusivity and the rate of collisional dissipation of the fluctuation kinetic energy, and successfully test the associated constitutive relations of the extended kinetic theory, i.e., a kinetic theory which includes the role of velocity correlations. We then phrase and numerically integrate a system of differential equations governing the flow, with suitably modified boundary conditions. We show a remarkable qualitative and quantitative agreement with the results of the discrete simulations. In particular, we study the effect of (i) the coefficient of collisional restitution, (ii) the imposed load and (iii) the bumpiness of the planes on the profiles of the hydrodynamic fields, the ratio of shear stress-to-pressure and the gap between the bumpy planes. Finally, we predict the critical value of the imposed load above which crystallization occurs, based on the value of the solid volume fraction near the boundaries obtained from the numerical solution of the kinetic theory. This notably reproduces what we observe in the discrete simulations.
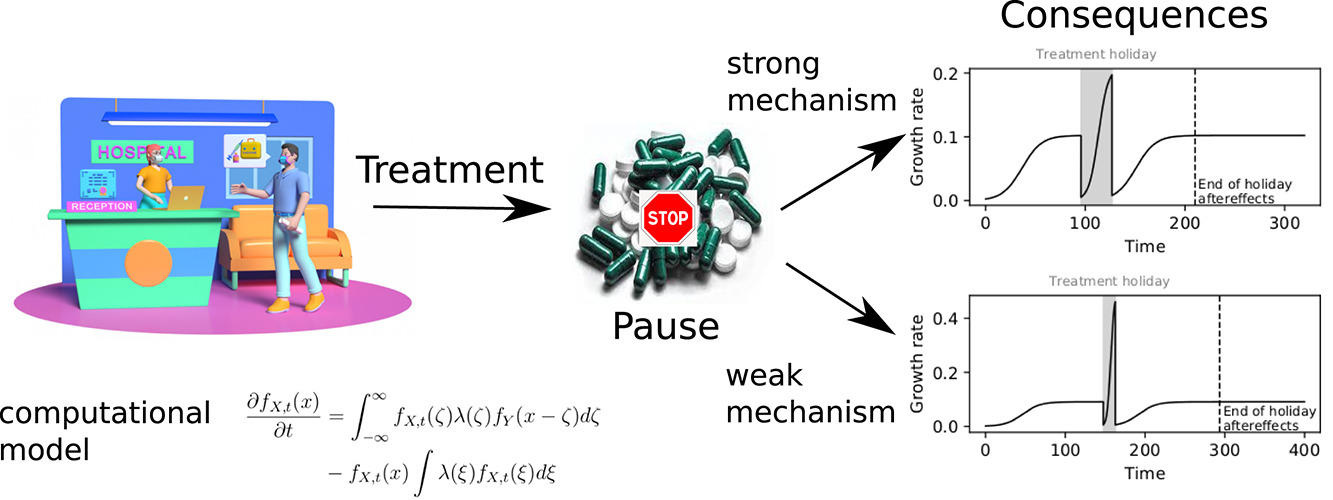
Interplay of mutations, alternate mechanisms, and treatment breaks in leukaemia: Understanding and implications studied with stochastic models
H. Jonathan G. Lindström, Astrid S. de Wijn, and Ran Friedman
Computers in Biology and Medicine 169 (2024) 107826.
H. Jonathan G. Lindström, Astrid S. de Wijn, and Ran Friedman
Computers in Biology and Medicine 169 (2024) 107826.
Bcr-Abl1 kinase domain mutations are the most prevalent cause of treatment resistance in chronic myeloid leukaemia (CML). Alternate resistance pathways nevertheless exist, and cell line experiments show certain patterns in the gain, and loss, of some of these alternate adaptations. These adaptations have clinical consequences when the tumour develops mechanisms that are beneficial to its growth under treatment, but slow down its growth when not treated. The results of temporarily halting treatment in CML have not been widely discussed in the clinic and there is no robust theoretical model that could suggest when such a pause in therapy can be tolerated. We constructed a dynamic model of how mechanisms such as Bcr-Abl1 overexpression and drug transporter upregulation evolve to produce resistance in cell lines, and investigate its behaviour subject to different treatment schedules, in particular when the treatment is paused (‘drug holiday’). Our study results suggest that the presence of additional resistance mechanisms creates an environment which favours mutations that are either preexisting or occur late during treatment. Importantly, the results suggest the existence of tumour drug addiction, where cancer cells become dependent on the drug for (optimal) survival, which could be exploited through a treatment holiday. All simulation code is available at github.
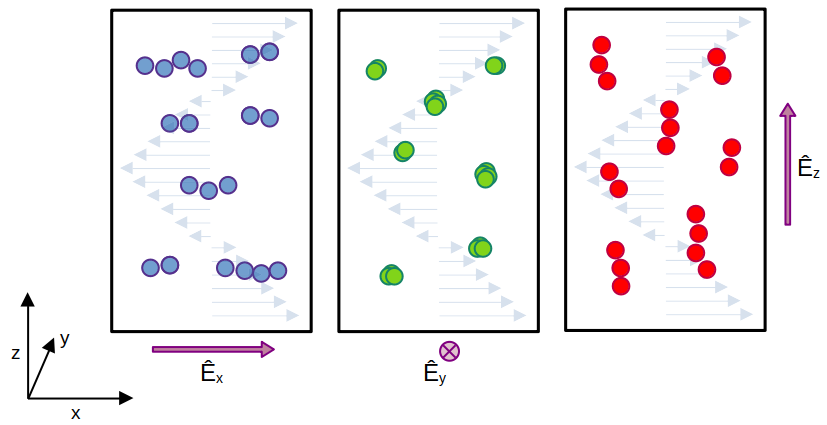
Anisotropy of field-controlled shear viscosity of dipolar fluids
Christopher D. Fjeldstad, Faezeh Pousaneh, Roberto E. Troncoso, and Astrid S. de Wijn
J. Stat. Mech. (2023) 123204.
Christopher D. Fjeldstad, Faezeh Pousaneh, Roberto E. Troncoso, and Astrid S. de Wijn
J. Stat. Mech. (2023) 123204.
We numerically study the anisotropic viscous response of dipolar hard spheres in the presence of an electric field. We find that the shear viscosity is sensitive to both the strength and orientation of the field relative to the shearing direction. The effect on the viscosity is strongest when the field is oriented in the shear gradient direction. We investigate the structure of the fluid in detail to explain the mechanism for the changes in viscosity, and show that the reorientation of chain-like clusters plays a crucial role. We show that the anisotropy arises from the polarization of the fluid induced by the field, leading to the orientation of chain-like clusters along the direction of the field.

Nanoscale tribological simulations of a semi-crystalline polymer
Robin Vacher
PhD thesis, Norwegian University of Science and Technology (2022).
Robin Vacher
PhD thesis, Norwegian University of Science and Technology (2022).
Many of the objects surrounding us are made of polymers. Those polymers are often used for their tribological properties, for example, shoes with rubber soles or car tyres. Polymers materials become more and more presents, and their frictional behaviour is often a significant issue. Macroscopic properties of those materials such as friction and wear have been intensely studied in the past. Those studies have shown that there are nontrivial effects in friction and wear specific to polymers, such as non-linearity and nontrivial temperature dependence. Much remains to be understood, as there are many additional complications in many realistic polymers that can affect the structure and friction, such as the strength of the interatomic interactions, cross-linking, or the presence of water and other contaminants. We want to identify and investigate some of the main mechanisms of semicrystalline polymer friction. In that regard, molecular dynamic simulations are used to create semi-crystalline solid polymer substrates at the nanoscopic scale. We modelled a friction force microscope experiment. The polymer tends to coaxially align and form a layered structure during rubbing simulations directly under the tip. Over time, the plastic deformation on and near the surface builds up, the friction decreases, and the polymers in the top layer align with each other in the sliding direction. A small amount of friction is often wanted in tribological systems because high friction is linked to high wear and large energy consumption. A way to reduce friction is to apply lubricants. We have put the focus of our study on graphene lubricant. One of our goals is to identify how adding a graphene layer helps reduce friction and wear. We found that the stiffness of the graphene membrane linked to the boundary condition has a substantial impact on the indentation depth. The surface profiles are directly affecting the friction. We investigate the emergence of the roughness of a polymer material. We found that by compressing a solid PVA substrate, the roughness of the polymer self-affinity continues to change with increasing strain.We associate this phenomenon with the viscoelastic properties of the polymer. While our simulations are for a specific polymer, the qualitative behaviour is likely to be general and present in other polymers.
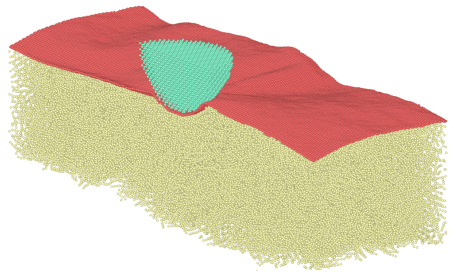
Nanoscale friction and wear of a polymer coated with graphene
Robin Vacher and Astrid S. de Wijn
Beilstein J. Nanotechnol. 2022, 13, 63–73. doi:10.3762/bjnano.13.4.
Robin Vacher and Astrid S. de Wijn
Beilstein J. Nanotechnol. 2022, 13, 63–73. doi:10.3762/bjnano.13.4.
Friction and wear of polymers at the nanoscale is a challenging problem due to the complex viscoelastic properties and structure. Using molecular dynamics simulations, we investigate how a graphene sheet on top of the semicrystalline polymer polyvinyl alcohol affects the friction and wear. Our setup is meant to resemble an AFM experiment with a silicon tip. We have used two different graphene sheets, namely an unstrained, flat sheet, and one that has been crumpled before being deposited on the polymer. The graphene protects the top layer of the polymer from wear and reduces the friction. The unstrained flat graphene is stiffer, and we find that it constrains the polymer chains and reduces the indentation depth.
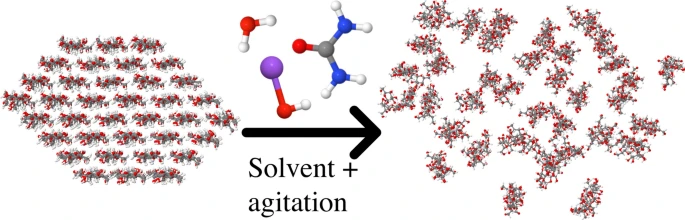
Computational study of the dissolution of cellulose into single chains: the role of the solvent and agitation
Eivind Bering, Jonathan Ø. Torstensen, Anders Lervik, and Astrid S. de Wijn
Cellulose (2022).
Eivind Bering, Jonathan Ø. Torstensen, Anders Lervik, and Astrid S. de Wijn
Cellulose (2022).
We investigate the dissolution mechanism of cellulose using molecular dynamics simulations in both water and a mixture solvent consisting of water with Na\(^+\), OH\(^-\) and urea. As a first computational study of its kind, we apply periodic external forces that mimic agitation of the suspension. Without the agitation, the bundles do not dissolve, neither in water nor solvent. In the solvent mixture the bundle swells with significant amounts of urea entering the bundle, as well as more water than in the bundles subjected to pure water. We also find that the mixture solution stabilizes cellulose sheets, while in water these immediately collapse into bundles. Under agitation the bundles dissolve more easily in the solvent mixture than in water, where sheets of cellulose remain that are bound together through hydrophobic interactions. Our findings highlight the importance of urea in the solvent, as well as the hydrophobic interactions, and are consistent with experimental results.
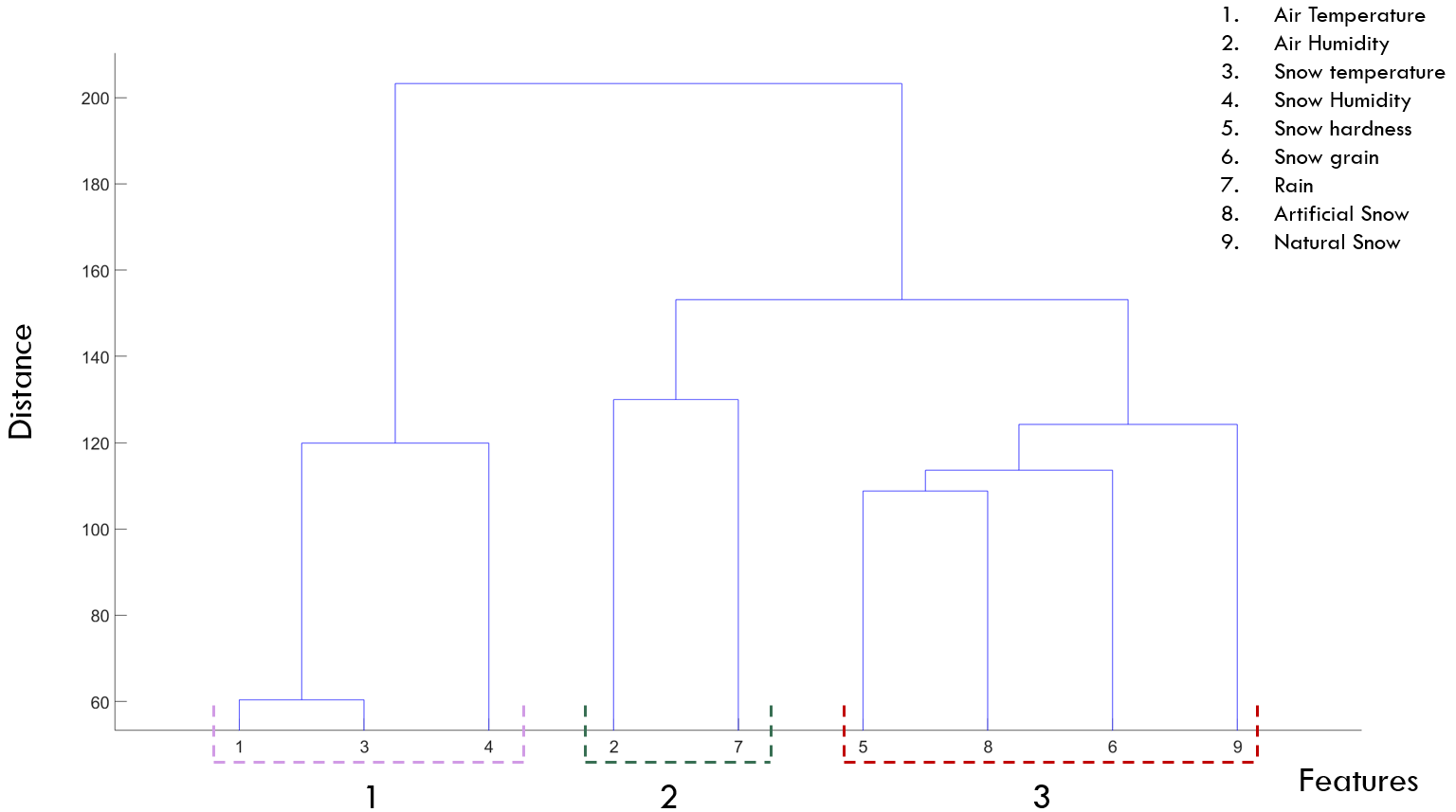
Identifying ski roughness using data driven approaches
Bassma Al-Jubouri and Astrid S. de Wijn
2021 IEEE International Conference on Systems, Man, and Cybernetics (SMC), 2021, pp. 1861-1868, doi: 10.1109/SMC52423.2021.9658737.
Bassma Al-Jubouri and Astrid S. de Wijn
2021 IEEE International Conference on Systems, Man, and Cybernetics (SMC), 2021, pp. 1861-1868, doi: 10.1109/SMC52423.2021.9658737.
In skiing sport, snow friction is a crucial factor in determining the ski roughness that can produce high speed and quick finishing time for a skier. However, snow friction is influenced by many factors associated with weather and snow conditions which affect the choice of the optimal ski’s roughness. This paper proposes an ensemble learning system that can accurately recommend the best ski roughness under different weather conditions. The data used in this study is a unique data set that has been collected from field tests and competitions. Though this data set recorded information about ski treatment and weather conditions over a 10-years period, it is affected by noise and outliers, and it has an imbalanced distribution in the ski roughness classes. This work addresses these challenges in the data by applying preprocessing techniques and class balancing strategies. Furthermore, correlation and clustering approaches are employed to identify redundancies in the data and to recognise the subsets of weather conditions that have the highest influence on the selection of the ski roughness. Using the resultant clusters, an ensemble system is introduced to recommend the most suitable skis roughness for a given weather condition. This system can be used as a guiding tool in skiing competitions to aid technicians in choosing the skis roughness. The results showed that air and snow temperatures as well as snow humidity have the highest impact on the choice of the ski roughness.
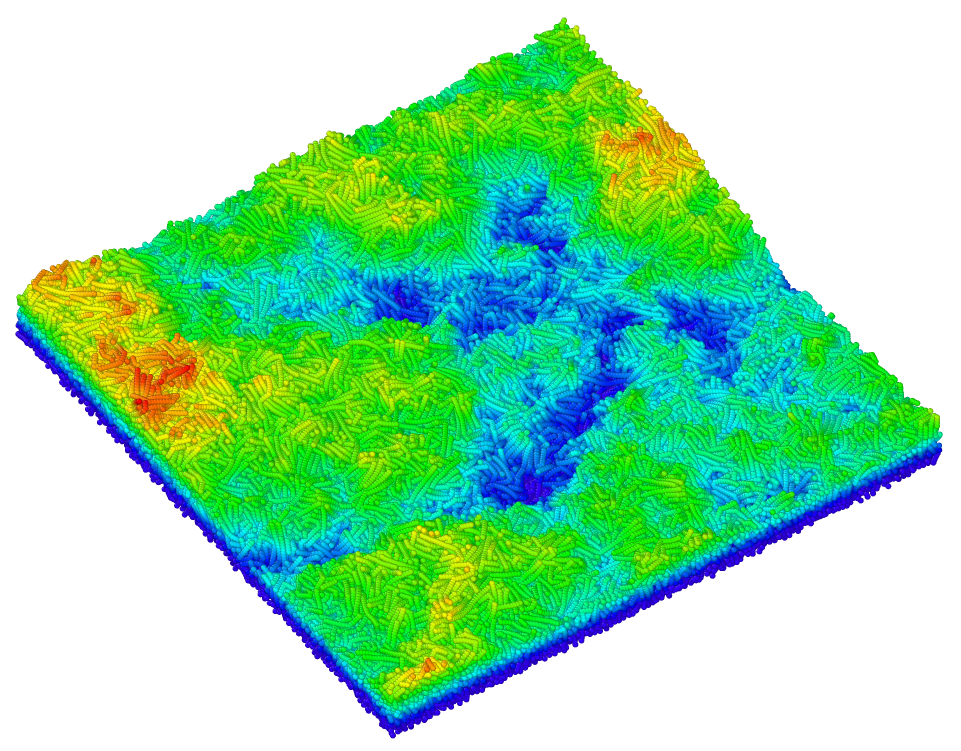
Molecular-Dynamics Simulations of the Emergence of Surface Roughness in a Polymer under Compression
Robin Vacher and Astrid S. de Wijn
Materials 2021, 14(23), 7327.
Robin Vacher and Astrid S. de Wijn
Materials 2021, 14(23), 7327.
Roughness of surfaces is both surprisingly ubiquitous on all length scales and extremely relevant practically. The appearance of multi-scale roughness has been linked to avalanches and plastic deformation in metals. However, other, more-complex materials have mechanisms of plasticity that are significantly different from those of metals. We investigated the emergence of roughness in a polymer under compression. We performed molecular-dynamics simulations of a slab of solid polyvinyl alcohol that was compressed bi-axially, and we characterised the evolution of the surface roughness. We found significantly different behaviour than what was previously observed in similar simulations of metals. We investigated the differences and argue that the visco-elasticity of the material plays a crucial role.

Stretching, breaking, and dissolution of polymeric nanofibres by computer experiments
Eivind Bering
PhD thesis, Norwegian University of Science and Technology (2021).
Eivind Bering
PhD thesis, Norwegian University of Science and Technology (2021).
Bundles of polymeric materials are ubiquitous and play essential roles in biological systems, and often display remarkable mechanical properties. With the never-ending experimental advances in control and manipulation of molecular properties on the nanometric level follows an increasing demand for a theoretical description that is valid at this scale. This regime of nano-scale bundles of small numbers of molecules has not been investigated much theoretically; here chain–chain interactions, surface effects, entropy, nonlinearities, and thermal fluctuations all play important roles. In my thesis, I present a broad exploration by molecular-dynamics simulations of single chains and bundles under external loading. Stretching and rearrangements of chains are investigated, as well as their breaking and dissolution.
Thermal effects and spontaneous frictional relaxation in atomically thin layered materials
J. Roadnight Sheehan, David Andersson, and Astrid S. de Wijn
Phys. Rev. B 103, 195441 (2021).
J. Roadnight Sheehan, David Andersson, and Astrid S. de Wijn
Phys. Rev. B 103, 195441 (2021).
We study the thermal effects on the frictional properties of atomically thin sheets. We simulate a simple model based on the Prandtl-Tomlinson model that reproduces the layer dependence of friction and strengthening effects seen in AFM experiments. We investigate sliding at constant speed as well as reversing direction. We also investigate contact aging: the changes that occur to the contact when the sliding stops completely. We compare the numerical results to analytical calculations based on Kramers rates. We find that there is a slower than exponential contact aging that weakens the contact and that we expect will be observable in experiments. We discuss the implications for sliding as well as aging experiments.
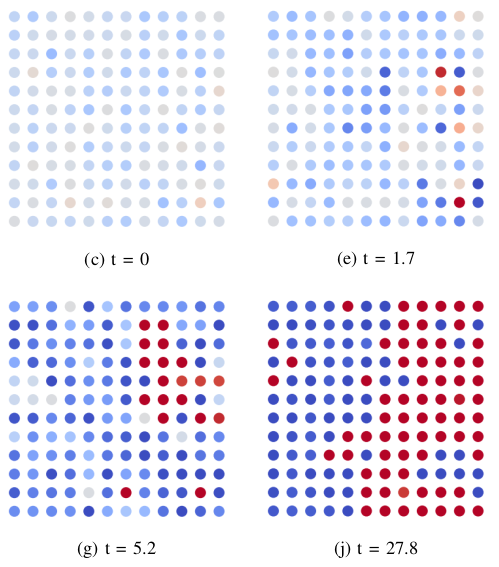
Dynamics of collective action to conserve a large common-pool resource
David Andersson, Sigrid Bratsberg, Andrew K. Ringsmuth, Astrid S. de Wijn
Scientific Reports volume 11, Article number: 9208 (2021).
David Andersson, Sigrid Bratsberg, Andrew K. Ringsmuth, Astrid S. de Wijn
Scientific Reports volume 11, Article number: 9208 (2021).
A pressing challenge for coming decades is sustainable and just management of large-scale common-pool resources including the atmosphere, biodiversity and public services. This poses a difficult collective action problem because such resources may not show signs that usage restraint is needed until tragedy is almost inevitable. To solve this problem, a sufficient level of cooperation with a pro-conservation behavioural norm must be achieved, within the prevailing sociopolitical environment, in time for the action taken to be effective. Here we investigate the transient dynamics of behavioural change in an agent-based model on structured networks that are also exposed to a global external influence. We find that polarisation emerges naturally, even without bounded confidence, but that for rationally motivated agents, it is temporary. The speed of convergence to a final consensus is controlled by the rate at which the polarised clusters are dissolved. This depends strongly on the combination of external influences and the network topology. Both high connectivity and a favourable environment are needed to rapidly obtain final consensus.

Hydrogen assisted crack growth in iron: a simulations approach
Jan Inge Hammer Meling
PhD thesis, Norwegian University of Science and Technology (2021).
Jan Inge Hammer Meling
PhD thesis, Norwegian University of Science and Technology (2021).
Hydrogen enhances crack growth, limiting the lifetime of industrial components. Despite the abundance of research into hydrogen enhanced crack growth and its proposed mechanisms, none of the models can explain all the observations. The effect of hydrogen on the growth of a sharp crack in a Fe-3wt%Si is an enhanced crack growth rate and a sharper crack. The higher the hydrogen gas pressure, the sharper the crack tip. Other observations include a reduced residual plastic zone. The objective of this thesis is to investigate how hydrogen may cause the enhanced crack growth rate and crack sharpening by implementing variants of the hydrogen mechanism HEDE and HELP. In this work, we show that hydrogen can activate mechanisms suggested by the literature, and the deciding factor is system-specific. Two different simulation models are used to investigate the hydrogen effect. The first is a study on a sharp crack simulated using 2D discrete dislocation dynamics. In the case of a sharp crack tip in Fe-3wt%Si, a reduction in the cohesive energy at the crack tip is the most critical for the crack growth and sharpening of the crack. The second study is a time-dependent microstructure evolution model of nanoindentation. In the case of nanoindentation, the reduction in the line energy of dislocations is the most prominent hydrogen effect. Both models emulate the hydrogen mechanisms through the defactant framework. The defactant framework describes the hydrogen influence as a reduction in the energy of defects as a function of the chemical potential of hydrogen. The hydrogen effect of reducing the line energy of dislocations does not replicate the sharpening of the crack. A reduction in the cohesive force, on the other hand, does. A lowered cleavage criterion due to decohesion at the crack tip results in less plasticity before a cleavage event. Activation of a cleavage event at a lower load replicates the experimental findings of lowered fracture toughness, enhanced crack growth rate and a sharper crack tip. However, this does not rule out the hydrogen effect on dislocations, as that hydrogen effect replicates the lowered pop-in seen in nanoindentation experiments. Hydrogen can promote several fracture types; intergranular, transgranular or void growth and coalescence. Which fracture type hydrogen promotes, depends on the loading conditions, charging conditions and the underlying microstructure. Hydrogen promotes local deformation where it accumulates, and different loading and charging conditions can change where and at what rate hydrogen is accumulating. The hydrogen effect should be linked to the charging conditions using the chemical potential of hydrogen. We suggest the best way forward is to assume the hydrogen effect is a hybrid of several mechanisms, and which is critical depends on the system.

Simple Models for Complex Nonequilibrium Problems in Nanoscale Friction and Network Dynamics
David Andersson
PhD thesis, Department of Physics, Stockholm University (2021).
David Andersson
PhD thesis, Department of Physics, Stockholm University (2021).
This doctoral thesis investigates three different topics: How friction evolves in atomically thin layered materials (2D materials); How social dynamics can be used to model grand scale common-pool resource games; Benchmarking of various image reconstruction algorithms in atomic force microscopy experiments. While these topics are diverse, they share being complex out-of-equilibrium systems. Furthermore, our approach to these topics will be the same: using simple models to obtain qualitative information about a system's dynamics. In the case of atomically thin layered materials, we will be expanding on the influential Prandtl-Tomlinson model and obtain an improved model constituting a substantial improvement in the theoretical description of friction in these systems. In the context of social dynamics, we will introduce a novel model representing a new approach to consensus rates on social networks in relation to society spanning coordination problems. For the image reconstruction project, our ambition is to investigate a new method for recreating free-energy surfaces based on AFM experiment, however, for this project only preliminary results are included.

Nanoscale Simulations of Wear and Viscoelasticity of a Semi-Crystalline Polymer
R. Vacher and Astrid S. de Wijn
Tribology Letters 69, 1-12 (2021).
R. Vacher and Astrid S. de Wijn
Tribology Letters 69, 1-12 (2021).
We investigate the underlying tribological mechanisms and running-in process of a semi-crystalline polymer using molecular-dynamics simulations. We subject a slab of simulated polyvinyl alcohol to a sliding contact asperity resembling a friction force microscope tip. We study the viscoelastic response of the polymer to the sliding and show both plastic and elastic contributions to the deformation, with their relative strength dependent on the temperature. As expected, the elastic deformation penetrates deeper into the surface than the plastic deformation. Directly under the tip, the polymer has a tendency to co-axially align and form a layered structure. Over time, the plastic deformation on and near the surface builds up, the friction decreases, and the polymers in the top layer align with each other in the sliding direction (conditioning).
A Legendre–Fenchel Transform for Molecular Stretching Energies
Eivind Bering, Dick Bedeaux, Signe Kjelstrup, Astrid S. de Wijn, Ivan Latella, and J. Miguel Rubi
Nanomaterials 2020, 10, 2355 (2020).
Eivind Bering, Dick Bedeaux, Signe Kjelstrup, Astrid S. de Wijn, Ivan Latella, and J. Miguel Rubi
Nanomaterials 2020, 10, 2355 (2020).
Single-molecular polymers can be used to analyze to what extent thermodynamics applies when the size of the system is drastically reduced. We have recently verified using molecular-dynamics simulations that isometric and isotensional stretching of a small polymer result in Helmholtz and Gibbs stretching energies, which are not related to a Legendre transform, as they are for sufficiently long polymers. This disparity has also been observed experimentally. Using molecular dynamics simulations of polyethylene-oxide, we document for the first time that the Helmholtz and Gibbs stretching energies can be related by a Legendre–Fenchel transform. This opens up a possibility to apply this transform to other systems which are small in Hill’s sense.
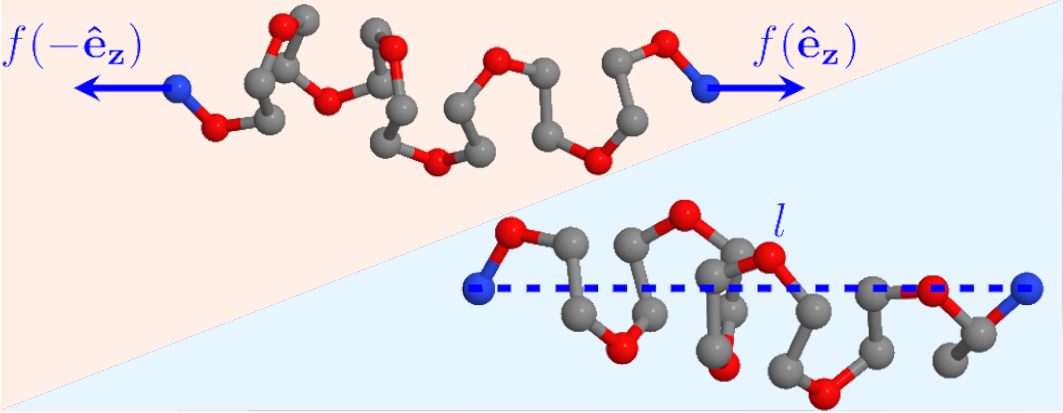
Entropy Production beyond the Thermodynamic Limit from Single-Molecule Stretching Simulations
Eivind Bering, Signe Kjelstrup, Dick Bedeaux, J. Miguel Rubi, and Astrid S. de Wijn
J. Phys. Chem. B 124, 40, 8909–8917 (2020).
Eivind Bering, Signe Kjelstrup, Dick Bedeaux, J. Miguel Rubi, and Astrid S. de Wijn
J. Phys. Chem. B 124, 40, 8909–8917 (2020).
Single-molecular systems are a test bed to analyze to what extent thermodynamics applies when the size of the system is drastically reduced. Isometric and isotensional single-molecule stretching experiments and their theoretical interpretations have shown the lack of a thermodynamic limit at those scales and the nonequivalence between their corresponding statistical ensembles. This disparity between thermodynamic results obtained in both experimental protocols can also be observed in entropy production, as previous theoretical results have shown. In this work, we present results from molecular dynamics simulations of stretching of a typical polymer, polyethylene-oxide, where this framework is applied to obtain friction coefficients associated with stretching at the two different statistical ensembles for two different system sizes, from which the entropy production follows. In the smallest system, they are different up to a factor of 2, and for the bigger system, the difference is smaller, as predicted. In this way, we provide numerical evidence that a thermodynamic description is still meaningful for the case of single-molecule stretching.
Kinetic theory and shear viscosity of dense dipolar hard sphere liquids
Faezeh Pousaneh and Astrid S. de Wijn
Phys. Rev. Lett. 124, 218004 (2020).
Faezeh Pousaneh and Astrid S. de Wijn
Phys. Rev. Lett. 124, 218004 (2020).
Transport properties of dense fluids are fundamentally challenging, because the powerful approaches of equilibrium statistical physics cannot be applied. Polar fluids compound this problem, because the long-range interactions preclude the use of a simple effect-diameter approach based solely on hard spheres. Here, we develop a kinetic theory for dipolar hard-sphere fluids that is valid up to high density. We derive a mathematical approximation for the radial distribution function at contact directly from the equation of state, and use it to obtain the shear viscosity. We also perform molecular-dynamics simulations of this system and extract the shear viscosity numerically. The theoretical results compare favorably to the simulations.
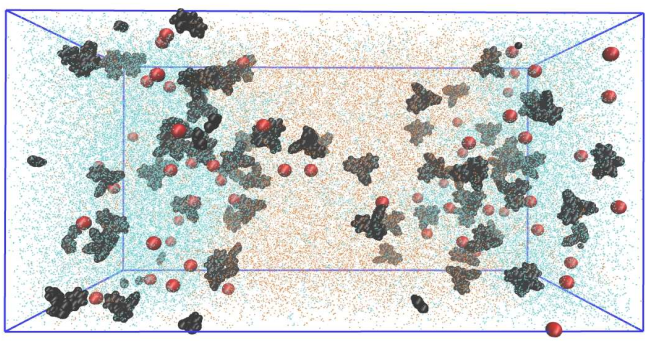
The Vanishing water/oil interface in the presence of antagonistic salt
Gudrun Glende, Astrid S. de Wijn, and Faezeh Pousaneh
J. Chem. Phys. 152, 124707 (2020).
Gudrun Glende, Astrid S. de Wijn, and Faezeh Pousaneh
J. Chem. Phys. 152, 124707 (2020).
Antagonistic salts are salts that consist of hydrophilic and hydrophobic ions. In a binary mixture of water and an organic solvent, these ions preferentially dissolve into different phases. We investigate the effect of an antagonistic salt, tetraphenylphosphonium chloride PPh4+Cl−, in a mixture of water and 2,6-lutidine by means of Molecular Dynamics (MD) simulations. For increasing concentrations of the salt, the two-phase region is shrunk and the interfacial tension in reduced, in contrast to what happens when a normal salt is added to such a mixture. The MD simulations allow us to investigate in detail the mechanism behind the reduction of the surface tension. We obtain the ion and composition distributions around the interface and determine the hydrogen bonds in the system and conclude that the addition of salt alters the hydrogen bonding.

Stretching and breaking of PEO nanofibres. A classical force field and ab initio simulation study
Eivind Bering and Astrid S. de Wijn
Soft Matter 16, 2736-2752 (2020).
Eivind Bering and Astrid S. de Wijn
Soft Matter 16, 2736-2752 (2020).
The burgeoning development of nanotechnology is allowing us to construct more and more nano-scale systems in the real world that used to only exist in computer simulations. Among them, nanofibres made of only a few aligned polymeric chains in particular might soon have important roles in nanofabrications as well as in nanomedicine. In this work, we present a broad exploration by computer simulations of elastic and inelastic properties of polyethylene-oxide (PEO) nanofibres under load. We cover the full range from unloaded fibres up to their breaking point, focusing on all features that arise from chain–chain interactions and collective behaviour of the chains. We employ both molecular dynamics (MD) simulations and density functional theory (DF). The classical force field is represented by a minimal reactive force field model, allowing for the breaking of covalent bonds. Density functional (DF) computations provide a benchmark to gauge and validate the empirical force field approach, and offer an intriguing view of the bundle chemical evolution after breaking. Force-field based MD is employed for the systematic investigation of bundles of up to 24 chains, and for a single bundle of 100 chains. Low-temperature results for bundles under moderate loading provide a size-dependent sequence of cross-sections, structures, cohesive energies and elastic properties. A remarkably high Young's modulus on the order of 100 GPa was estimated with DF and MD, explained by the semi-crystalline state of the fibres giving mechanical properties comparable to those of carbon nanotubes and of graphene. Breaking is investigated by simulations with constant strain rate or constant stress. The bundle breaks whenever the potential energy is raised above its metastability range, but also below that limit due to creep activated by thermal fluctuations. A Kramer's-type approximation for the rate of chain breaking is proposed and compared to simulation data.
News and Views | Why surface roughness is similar at different scales
Astrid S. de Wijn
Nature 578, 366-367 (2020).
Astrid S. de Wijn
Nature 578, 366-367 (2020).
Most surfaces are rough at many length scales. Simulations show that this characteristic originates at the atomic level in metal-based materials when smooth blocks of these materials are compressed.
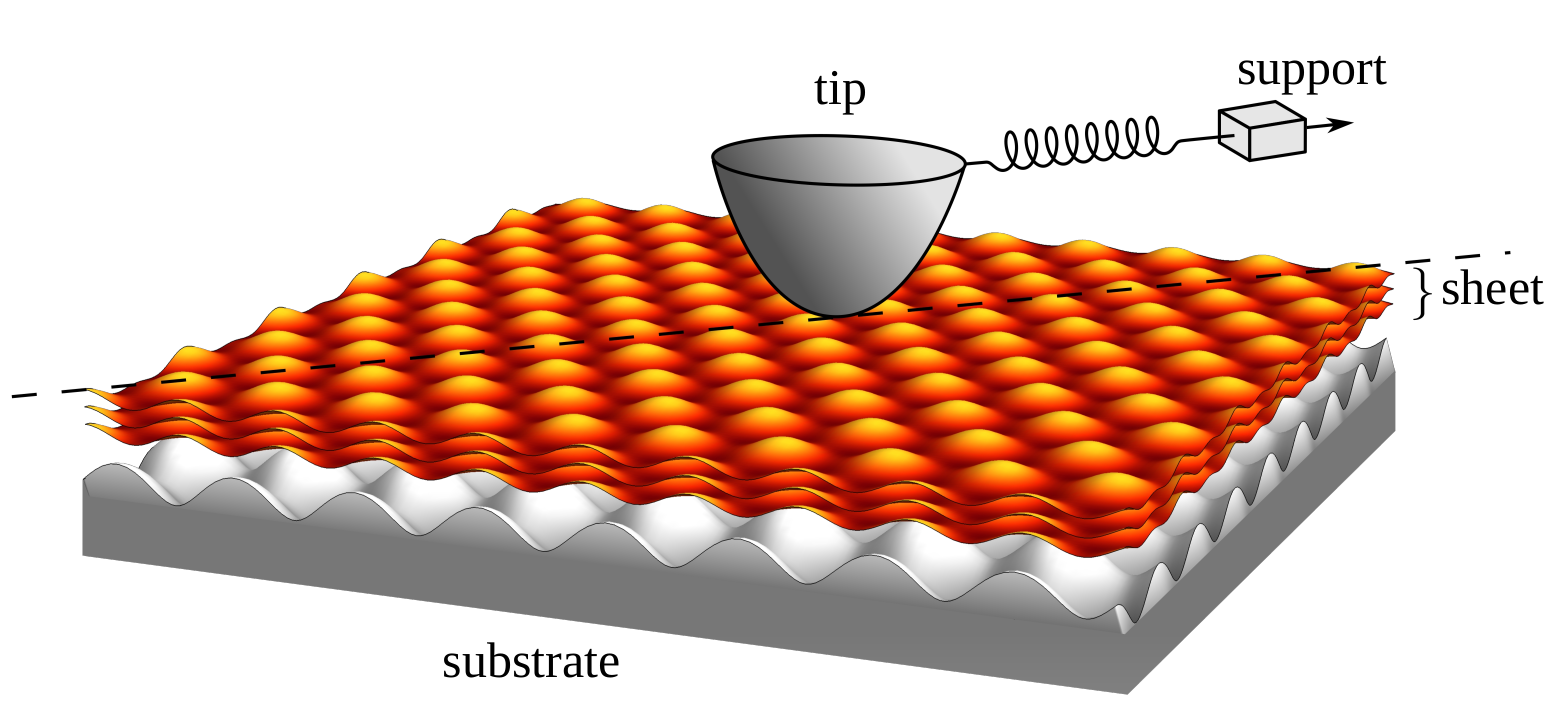
Understanding the friction of atomically thin layered materials
David Andersson and Astrid S. de Wijn
Nature Communications 11, 420 (2020).
David Andersson and Astrid S. de Wijn
Nature Communications 11, 420 (2020).
Friction is a ubiquitous phenomenon that greatly affects our everyday lives and is responsible for large amounts of energy loss in industrialised societies. Layered materials such as graphene have interesting frictional properties and are often used as (additives to) lubricants to reduce friction and protect against wear. Experimental Atomic Force Microscopy studies and detailed simulations have shown a number of intriguing effects such as frictional strengthening and dependence of friction on the number of layers covering a surface. Here, we propose a simple, fundamental, model for friction on thin sheets. We use our model to explain a variety of seemingly contradictory experimental as well as numerical results. This model can serve as a basis for understanding friction on thin sheets, and opens up new possibilities for ultimately controlling their friction and wear protection.
Shear viscosity of pseudo hard-spheres
Faezeh Pousaneh and Astrid S. de Wijn
Molecular Physics 118 1622050 (2020).
Faezeh Pousaneh and Astrid S. de Wijn
Molecular Physics 118 1622050 (2020).
We present molecular dynamics simulations of pseudo hard sphere fluid (generalized WCA poten- tial with exponents (50, 49) proposed by Jover et al. [J. Chem. Phys 137, (2012)] using GROMACS package. The equation of state and radial distribution functions at contact are obtained from simu- lations and compared to the available theory of true hard spheres (HS) and available data on pseudo hard spheres. The comparison shows agreements with data by Jover et al. and the Carnahan–Starling equation of HS. The shear viscosity is obtained from the simulations and compared to the Enskog expression and previous HS simulations. It is demonstrated that the PHS potential reproduces the HS shear viscosity accurately.

Friction vs. Area Scaling of Superlubric NaCl-Particles on Graphite
Felix Hartmuth, Dirk Dietzel, Astrid S. de Wijn, and André Schirmeisen
Lubricants 2019 7(8), 66 (2019).
Felix Hartmuth, Dirk Dietzel, Astrid S. de Wijn, and André Schirmeisen
Lubricants 2019 7(8), 66 (2019).
Structural lubricity is an intriguing tribological concept, where extremely low friction is anticipated, if two surfaces in relative motion do not share the same lattice structure and consequently instabilities originating from interlocking surface potentials are strongly reduced. Currently, the challenges related to the phenomenon of structural lubricity are considered to be twofold. On one hand, experimental systems suitable for showing structural lubricity must be identified, while at the same time, it is also crucial to understand the intricate details of interface interaction. Here, we introduce a new material combination, namely NaCl-particles on highly oriented pyrolithic graphite (HOPG), where the nanoparticles coalesce under the influence of ambient humidity. Our experiments reveal that the interfacial friction can be described by the concept of structural lubricity despite the seemingly unavoidable contamination of the interface. By systematically analyzing the friction versus area scaling, this unlikely candidate for structural lubricity then shows two separate friction branches, with distinct differences of the friction versus area scaling. The exact tribological behavior of the nanoparticles can ultimately be understood by a model that considers the influence of nanoparticle preparation on the interface conditions. By taking into account an inevitable water layer at the interface between particle and substrate that can exist in different crystalline configurations all friction phenomena observed in the experiments can be understood.
Stochastic modelling of tyrosine kinase inhibitor rotation therapy in chronic myeloid leukaemia
H. Jonathan G. Lindström, Astrid S. de Wijn, and Ran Friedman
BMC Cancer 19, 508 (2019).
H. Jonathan G. Lindström, Astrid S. de Wijn, and Ran Friedman
BMC Cancer 19, 508 (2019).
Background: Resistance towards targeted cancer treatments caused by single nucleotide variations is a major issue in many malignancies. Currently, there are a number of available drugs for chronic myeloid leukaemia (CML), which are overcome by different sets of mutations. The main aim of this study was to explore if it can be possible to exploit this and create a treatment protocol that outperforms each drug on its own.
Methods: We present a computer program to test different treatment protocols against CML, based on available resistance mutation growth data. The evolution of a relatively stable pool of cancer stem cells is modelled as a stochastic process, with the growth of cells expressing a tumourigenic protein (here, Abl1) and any emerging mutants determined principally by the drugs used in the therapy.
Results: There can be some benefit to Bosutinib-Ponatinib rotation therapy even if the mutation status is unknown, whereas Imatinib-Nilotinib rotation is unlikely to improve the outcomes. Furthermore, an interplay between growth inhibition and selection effects generates a non-linear relationship between drug doses and the risk of developing resistance.
Conclusions: Drug rotation therapy might be able to delay the onset of resistance in CML patients without costly ongoing observation of mutation status. Moreover, the simulations give credence to the suggestion that lower drug concentrations may achieve better results following major molecular response in CML.
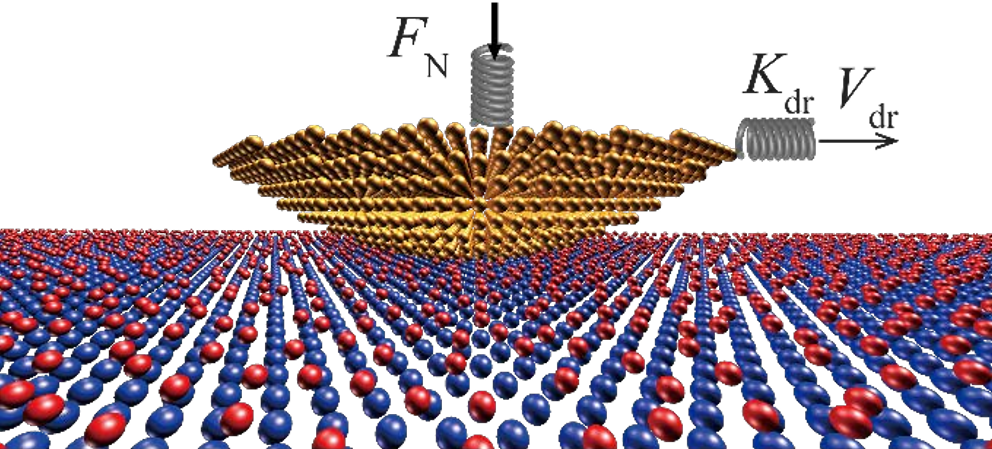
Atomic-scale sliding friction on a contaminated surface
Wengen Ouyang, Astrid S. de Wijn, and Michael Urbakh
Nanoscale 10, 6375-6381 (2018).
Wengen Ouyang, Astrid S. de Wijn, and Michael Urbakh
Nanoscale 10, 6375-6381 (2018).
Using non-equilibrium molecular dynamic simulations, we investigate the effect of adsorbates on nanoscopic friction. We find that the interplay between different channels of energy dissipation at the frictional interface may lead to non-monotonic dependence of the friction force on the adsorbate surface coverage and to strongly nonlinear variation of friction with normal load (non-Amontons’ behavior). Our simulations suggest that the key parameter controlling the variation of friction force with the normal load, surface coverage and temperature is the time-averaged number of adsorbates confined between the tip and the substrate. Three different regimes of temperature dependence of friction in the presence of adsorbates are predicted. Our findings point on new ways to control friction on contaminated surfaces.

Friction Fluctuations of Gold Nanoparticles in the Superlubric Regime
Dirk Dietzel, Astrid S. de Wijn, Matthias Vorholzer, and Andre Schirmeisen
Nanotechnology 29, 155702 (2018).
Dirk Dietzel, Astrid S. de Wijn, Matthias Vorholzer, and Andre Schirmeisen
Nanotechnology 29, 155702 (2018).
Superlubricity, or alternatively termed structural (super)lubrictiy, is a concept where ultra-low friction is expected at the interface between sliding surfaces if these surfaces are incommensurate and thus unable to interlock. In this work, we now report on sudden, reversible, friction changes that have been observed during AFM-based nanomanipulation experiments of gold nanoparticles sliding on highly oriented pyrolythic graphite. These effects can be explained by rotations of the gold nanoparticles within the concept of structural superlubricity, where the occurrence of ultra-low friction can depend extremely sensitively on the relative orientation between the slider and the substrate. From our theoretical simulations it will become apparent how even miniscule magnitudes of rotation are compatible to the observed effects and how size and shape of the particles can influence the dependence between friction and relative orientation.
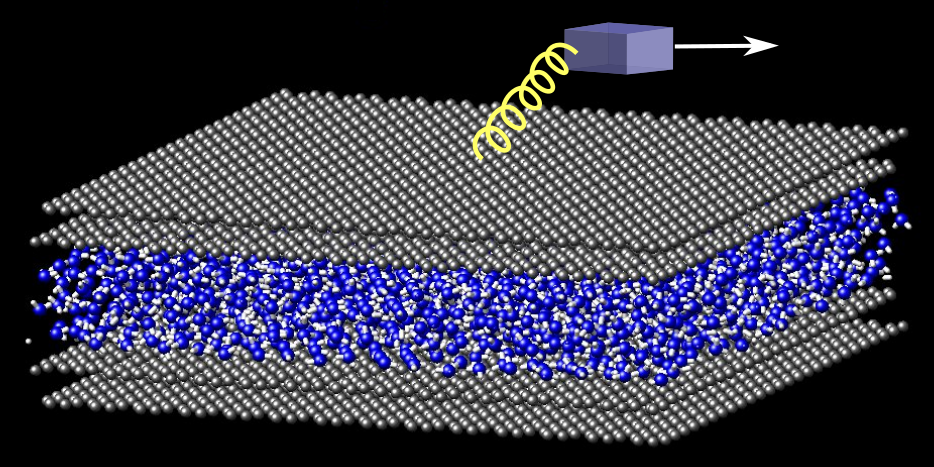
How square ice helps lubrication
Astrid S. de Wijn and Lars G. M. Pettersson
Phys. Rev. B 95, 165433 (2017).
Astrid S. de Wijn and Lars G. M. Pettersson
Phys. Rev. B 95, 165433 (2017).
In the context of friction we use atomistic molecular-dynamics simulations to investigate water confined between graphene sheets over a wide range of pressures. We find that thermal equilibration of the confined water is hindered at high pressures. We demonstrate that, under the right conditions, square ice can form in an asperity, and that it is similar to cubic ice VII and ice X. We simulate sliding of atomically flat graphite on the square ice and find extremely low friction due to structural superlubricity. The conditions needed for square ice to form correspond to low sliding speeds, and we suggest that the ice observed in experiments of friction on wet graphite is of this type.
News and Views | Nanoscience: Flexible graphene strengthens friction
Astrid S. de Wijn
Nature 539, 502–503 (2016).
Astrid S. de Wijn
Nature 539, 502–503 (2016).
Previous observations showed that friction on graphene increases gradually when a probe starts to slide across the material's surface. Simulations now reveal that this effect is related to bending of the graphene sheet. See Letter Nature 539, page 541
Imaging high-speed friction at the nanometer scale
Per-Anders Thorén, Astrid S. de Wijn, Riccardo Borgani, Daniel Forchheimer, and David B. Haviland
Nature Communications 7, 13836 (2016).
Per-Anders Thorén, Astrid S. de Wijn, Riccardo Borgani, Daniel Forchheimer, and David B. Haviland
Nature Communications 7, 13836 (2016).
Friction is a complicated phenomenon involving nonlinear dynamics at different length and time scales. The microscopic origin of friction is poorly understood, due in part to a lack of methods for measuring the force on a nanometer-scale asperity sliding at velocity of order cm/s. Dispite enormous advances in experimental technique this combination of small length scale and high velocity remained illusive. Here we present a technique for rapidly measuring the frictional forces on a single asperity (an AFM tip) over a velocity range from zero to several cm/s. At each image pixel we obtain the velocity dependence of both conservative and dissipative forces, revealing the transition from stick-slip to a smooth sliding friction. We explain measurements on graphite using a modified Prandtl-Tomlinson model that takes into account the damped elastic deformation of the asperity. With its significant improvement in force sensitivity and very small sliding amplitude, our method enables rapid and detailed surface mapping of the full velocity-dependence of frictional forces to sub 10 nm spatial resolution.
Emergent friction in two-dimensional Frenkel-Kontorova models
Jesper Norell, Annalisa Fasolino, Astrid S. de Wijn
Phys. Rev. E 94, 023001 (2016).
Jesper Norell, Annalisa Fasolino, Astrid S. de Wijn
Phys. Rev. E 94, 023001 (2016).
Simple models for friction are typically one-dimensional, but real interfaces are two-dimensional. We investigate the effects of the second dimension on static and dynamic friction by using the Frenkel-Kontorova (FK) model. We study the two most straightforward extensions of the FK model to two dimensions and simulate both the static and dynamic properties. We show that the behavior of the static friction is robust and remains similar in two dimensions for physically reasonable parameter values. The dynamic friction, however, is strongly influenced by the second dimension and the accompanying additional dynamics and parameters introduced into the models. We discuss our results in terms of the thermal equilibration and phonon dispersion relations of the lattices, establishing a physically realistic and suitable two-dimensional extension of the FK model. We find that the presence of additional dissipation channels can increase the friction and produces significantly different temperature dependence when compared to the one-dimensional case. We also briefly study the anisotropy of the dynamic friction and show highly nontrivial effects, including that the friction anisotropy can lead to motion in different directions depending on the value of the initial velocity.

Collective superlubricity of graphene flakes
Merel M. van Wijk, Astrid S. de Wijn, and Annalisa Fasolino
J. Phys.: Condens. Matter 28 134007 (2016).
Merel M. van Wijk, Astrid S. de Wijn, and Annalisa Fasolino
J. Phys.: Condens. Matter 28 134007 (2016).
We investigate solid lubrication of graphene and graphene flakes using atomistic molecular-dynamics simulations. We find that graphene flakes yield lower friction than graphene as a result of a collective mechanism that emerges from the independent behaviour of the flakes. By freezing out different degrees of freedom of the flakes, we are able to attribute the low friction to non-simultaneous slipping of the individual flakes. We also compare the results of the atomistic simulations to those of a simplified two-dimensional model and find that the behaviour of the latter is strongly dependent on parameters, which emerge naturally from the atomistic simulations.
Preface to the special section on nano- and mesoscale friction
Eran Bouchbinder, Adam S. Foster, Oğuzhan Gürlü, Ernst Meyer, Susan Perkin, Andre Schirmeisen and Astrid S. de Wijn
J. Phys.: Condens. Matter 28 130301 (2016).
Eran Bouchbinder, Adam S. Foster, Oğuzhan Gürlü, Ernst Meyer, Susan Perkin, Andre Schirmeisen and Astrid S. de Wijn
J. Phys.: Condens. Matter 28 130301 (2016).
Effects of molecule anchoring and dispersion on nanoscopic friction under electrochemical control
A. S. de Wijn, A. Fasolino, A. E. Filippov, and M. Urbakh
J. Phys.: Condens. Matter 28, 105001 (2016).
A. S. de Wijn, A. Fasolino, A. E. Filippov, and M. Urbakh
J. Phys.: Condens. Matter 28, 105001 (2016).
The application of electric fields is a promising strategy for in situ control of friction. While there have recently been many experimental studies on friction under the influence of electric fields, theoretical understanding is very limited. Recently, we introduced a simple theoretical model for friction under electrochemical conditions that focused on the interaction of a force microscope tip with adsorbed molecules whose orientation was dependent on the applied electric field. Here we focus on the effects of anchoring of the molecules on friction. We show that anchoring affects the intensity and width of the peak in the friction that occurs near a reorientation transition of adsorbed molecules, and explain this by comparing the strength of molecule–molecule and molecule–tip interactions. We derive a dispersion relation for phonons in the layer of adsorbed molecules and demonstrate that it can be used to understand important features of the frictional response.
Chaotic properties of spin lattices near second-order phase transitions
A. S. de Wijn, B. Hess, and B. V. Fine
Phys. Rev. E 92, 062929 (2015).
A. S. de Wijn, B. Hess, and B. V. Fine
Phys. Rev. E 92, 062929 (2015).
We perform a numerical investigation of the Lyapunov spectra of chaotic dynamics in lattices of classical spins in the vicinity of second-order ferromagnetic and antiferromagnetic phase transitions. On the basis of this investigation, we identify a characteristic of the shape of the Lyapunov spectra, the "G-index", which exhibits a sharp peak as a function of temperature at the phase transition, provided the order parameter is capable of sufficiently strong dynamic fluctuations. As a part of this work, we also propose a general numerical algorithm for determining the temperature in many-particle systems, where kinetic energy is not defined.
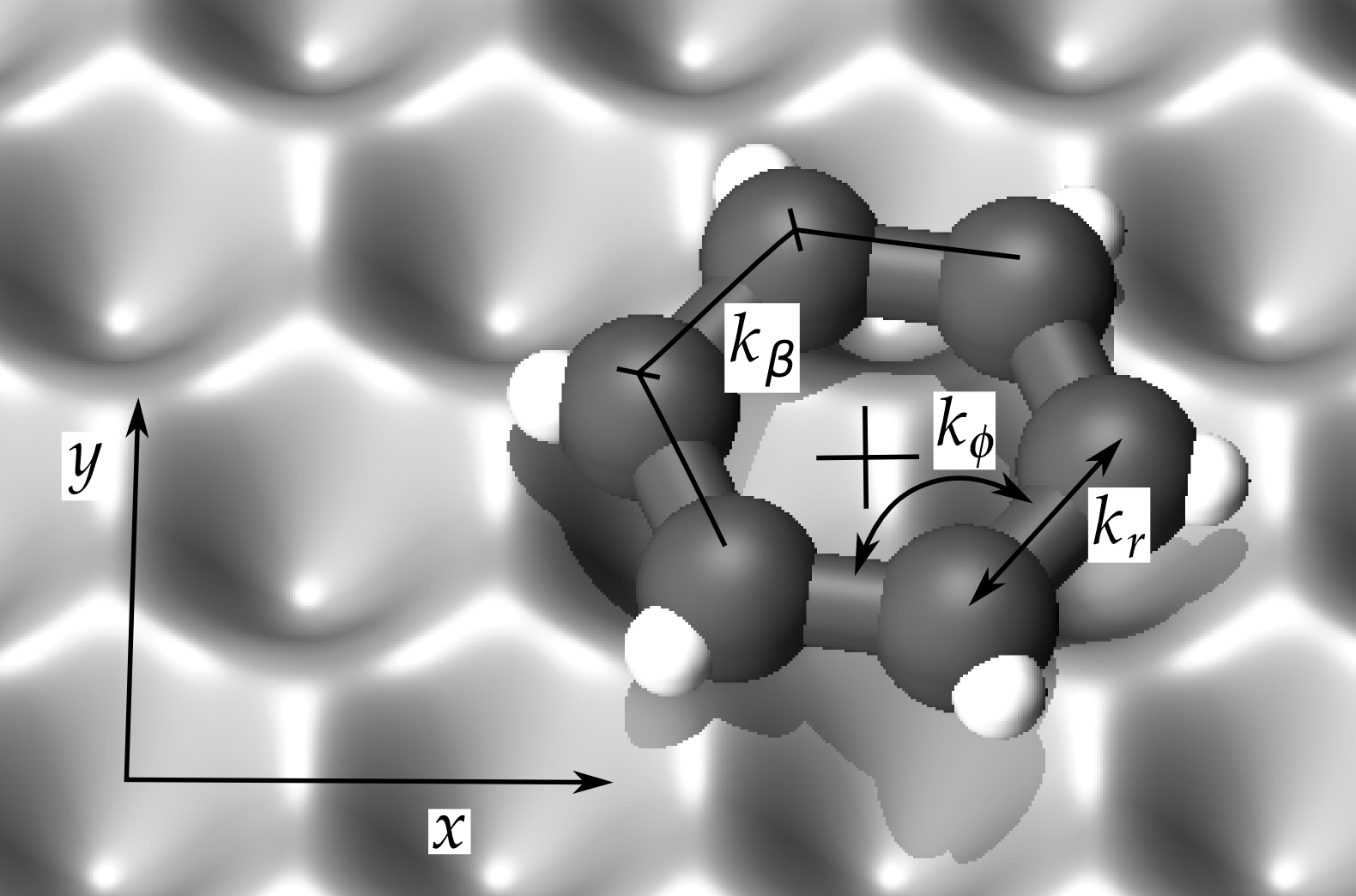
Understanding and controlling regime switching in molecular diffusion
S. Hallerberg and A. S. de Wijn
Phys. Rev. E 90, 062901 (2014).
S. Hallerberg and A. S. de Wijn
Phys. Rev. E 90, 062901 (2014).
Diffusion can be strongly affected by ballistic flights (long jumps) as well as long-lived sticking trajectories (long sticks). Using statistical inference techniques in the spirit of Granger causality, we investigate the appearance of long jumps and sticks in molecular-dynamics simulations of diffusion in a prototype system, a benzene molecule on a graphite substrate. We find that specific fluctuations in certain, but not all, internal degrees of freedom of the molecule can be linked to either long jumps or sticks. Furthermore, by changing the prevalence of these predictors with an outside influence, the diffusion of the molecule can be controlled. The approach presented in this proof of concept study is very generic, and can be applied to larger and more complex molecules. Additionally, the predictor variables can be chosen in a general way so as to be accessible in experiments, making the method feasible for control of diffusion in applications. Our results also demonstrate that data-mining techniques can be used to investigate the phase-space structure of high-dimensional nonlinear dynamical systems.
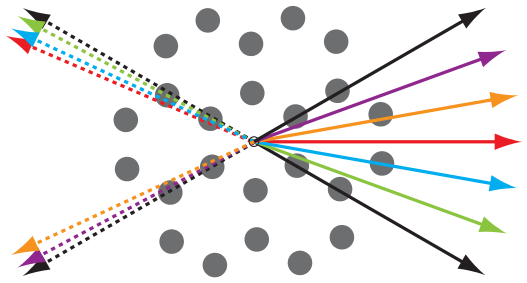
Preferential sliding directions on graphite
Balakrishna S.G., Astrid S. de Wijn, and Roland Bennewitz
Phys. Rev. B 89, 245440 (2014).
Balakrishna S.G., Astrid S. de Wijn, and Roland Bennewitz
Phys. Rev. B 89, 245440 (2014).
The anisotropy of friction on graphitic surfaces is investigated by a combined friction force microscopy and modeling study. Friction vectors deviate up to 15° from pulling directions. The strongest deviations are found for pulling directions which lie almost along one zigzag direction of the honeycomb structure, the preferred sliding direction on graphite surfaces and epitaxial graphene grown on SiC(0001). Atomic stick-slip events along and across molecular rows determine direction and magnitude of friction. Simulation and modeling reveal the role of temperature and of the two-dimensional character of the surface potential for the friction anisotropy.
A criterion for condensation in kinetically constrained one-dimensional transport models
D. M. Miedema, Astrid S. de Wijn, and Peter Schall
Phys. Rev. E 89, 062812 (2014).
D. M. Miedema, Astrid S. de Wijn, and Peter Schall
Phys. Rev. E 89, 062812 (2014).
We study condensation in one-dimensional transport models with a kinetic constraint. The kinetic constraint results in clustering of immobile vehicles; these clusters can grow to macroscopic condensates, indicating the onset of dynamic phase separation between free flowing and arrested traffic. We investigate analytically the conditions under which this occurs, and derive a necessary and sufficient criterion for phase separation. This criterion is applied to the well-known Nagel-Schreckenberg model of traffic flow to analytically investigate the existence of dynamic condensates. We find that true condensates occur only when acceleration out of jammed traffic happens in a single time step, in the limit of strong overbraking. Our predictions are further verified with simulation results on the growth of arrested clusters. These results provide analytic understanding of dynamic arrest and dynamic phase separation in one-dimensional traffic and transport models.
Absence of exponential sensitivity to small perturbations in nonintegrable systems of spins 1/2
Boris V. Fine, Tarek A. Elsayed, Chahan M. Kropf, and Astrid S. de Wijn
Phys. Rev. E 89, 012923 (2014).
Boris V. Fine, Tarek A. Elsayed, Chahan M. Kropf, and Astrid S. de Wijn
Phys. Rev. E 89, 012923 (2014).
We show that nonintegrable lattices of spins 1/2, which are often considered to be chaotic, do not exhibit the basic property expected for classical chaotic systems, namely, exponential sensitivity to small perturbations. Specifically, we compare the responses of chaotic lattices of classical spins and nonintegrable lattices of spins 1/2 to imperfect reversal of spin dynamics known as Loschmidt echo. In the classical case, Loschmidt echoes exhibit exponential sensitivity to small perturbations. This sensitivity is controlled by twice the value of the largest Lyapunov exponent of the underlying chaotic dynamics. In the case of spins 1/2, Loschmidt echoes are only power-law sensitive to small perturbations. The above findings imply that it is impossible to define Lyapunov exponents for translationally invariant lattices of spins 1/2 even in the macroscopic limit. The power-law sensitivity of spin 1/2 lattices to small perturbations is predicted to be measurable in nuclear magnetic resonance experiments. The above findings are encouraging for the efforts to create quantum simulators.
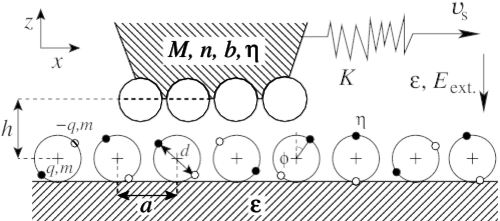
Nanoscopic friction under electrochemical control
A. S. de Wijn, A. Fasolino, A. Filippov, and M. Urbakh
Phys. Rev. Lett. 112, 055502 (2014).
A. S. de Wijn, A. Fasolino, A. Filippov, and M. Urbakh
Phys. Rev. Lett. 112, 055502 (2014).
We propose a theoretical model of friction under electrochemical conditions focusing on the interaction of a force microscope tip with adsorbed polar molecules of which the orientation depends on the applied electric field. We demonstrate that the dependence of friction force on the electric field is determined by the interplay of two channels of energy dissipation: (i) the rotation of dipoles and (ii) slips of the tip over potential barriers. We suggest a promising strategy to achieve a strong dependence of nanoscopic friction on the external field based on the competition between long range electrostatic interactions and short range chemical interactions between tip and adsorbed polar molecules.
Reconstruction of tip-surface interactions in multimodal intermodulation atomic force microscopy
Stanislav S. Borysov, Daniel Platz, Astrid S. de Wijn, Daniel Forchheimer, Eric A. Tolen, Alexander V. Balatsky, and David B. Haviland
Phys. Rev. B 88, 115405 (2013).
Stanislav S. Borysov, Daniel Platz, Astrid S. de Wijn, Daniel Forchheimer, Eric A. Tolen, Alexander V. Balatsky, and David B. Haviland
Phys. Rev. B 88, 115405 (2013).
We propose a theoretical framework for reconstructing tip-surface interactions using the intermodulation technique when more than one eigenmode is required to describe the cantilever motion. Two particular cases of bimodal motion are studied numerically: one bending and one torsional mode, and two bending modes. We demonstrate the possibility of accurate reconstruction of a two-dimensional conservative force field for the former case, while dissipative forces are studied for the latter.
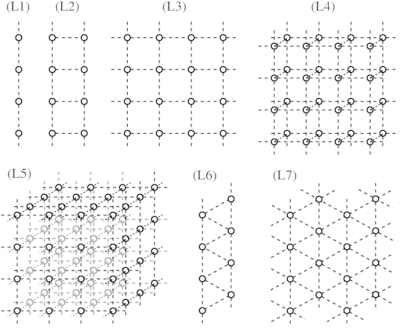
Lyapunov instabilities in lattices of interacting classical spins at infinite temperature
A. S. de Wijn, B. Hess, and B. V. Fine
J. Phys. A: Math. Theor. 46, 254012 (2013).
A. S. de Wijn, B. Hess, and B. V. Fine
J. Phys. A: Math. Theor. 46, 254012 (2013).
We numerically investigate Lyapunov instabilities for one-, two- and three-dimensional lattices of interacting classical spins at infinite temperature. We obtain the largest Lyapunov exponents for a very large variety of nearest-neighbor spin-spin interactions and complete Lyapunov spectra in a few selected cases. All Lyapunov spectra have weakly convex shapes and do not exhibit the Lyapunov modes observed for gases of hard-core particles. We investigate the dependence of the largest Lyapunov exponents and whole Lyapunov spectra on the lattice size and find that both quickly become size-independent. Finally, we analyze the dependence of the largest Lyapunov exponents on the anisotropy of spin-spin interaction with the particular focus on the difference between bipartite and nonbipartite lattices.
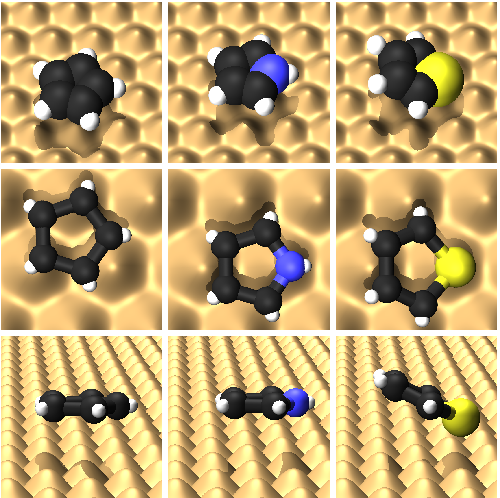
Atomic scale friction of molecular adsorbates during diffusion
B. A. J. Lechner¹, A. S. de Wijn¹, H. Hedgeland, A. P. Jardine, B. J. Hinch, W. Allison, and J. Ellis [¹joint first authors]
J. Chem. Phys. 138, 194710 (2013).
B. A. J. Lechner¹, A. S. de Wijn¹, H. Hedgeland, A. P. Jardine, B. J. Hinch, W. Allison, and J. Ellis [¹joint first authors]
J. Chem. Phys. 138, 194710 (2013).
Experimental observations suggest that molecular adsorbates exhibit a larger friction coefficient than atomic species of comparable mass, yet the origin of this increased friction is not well understood. We present a study of the microscopic origins of friction experienced by molecular adsorbates during surface diffusion. Helium spin-echo measurements of a range of five-membered aromatic molecules, cyclopentadienyl (Cp), pyrrole and thiophene, on a copper(111) surface are compared with molecular dynamics simulations of the respective systems. The adsorbates have different chemical interactions with the surface and differ in bonding geometry, yet the measurements show that the friction is greater than 2/ps for all these molecules. We demonstrate that the internal and external degrees of freedom of these adsorbate species are a key factor in the underlying microscopic processes and identify the rotation modes as the ones contributing most to the total measured friction coefficient.
Criticality in Dynamic Arrest: Correspondence between Glasses and Traffic
A. S. de Wijn, D. M. Miedema, B. Nienhuis, and P. Schall
Phys. Rev. Lett. 109, 228001 (2012).
A. S. de Wijn, D. M. Miedema, B. Nienhuis, and P. Schall
Phys. Rev. Lett. 109, 228001 (2012).
Dynamic arrest is a general phenomenon across a wide range of dynamic systems including glasses, traffic flow, and dynamics in cells, but the universality of dynamic arrest phenomena remains unclear. We connect the emergence of traffic jams in a simple traffic flow model directly to the dynamic slowing down in kinetically constrained models for glasses. In kinetically constrained models, the formation of glass becomes a true (singular) phase transition in the limit T→0. Similarly, using the Nagel-Schreckenberg model to simulate traffic flow, we show that the emergence of jammed traffic acquires the signature of a sharp transition in the deterministic limit p→1, corresponding to overcautious driving. We identify a true dynamic critical point marking the onset of coexistence between free flowing and jammed traffic, and demonstrate its analogy to the kinetically constrained glass models. We find diverging correlations analogous to those at a critical point of thermodynamic phase transitions.
The effect of temperature and velocity on superlubricity
Joost A. van den Ende, Astrid S. de Wijn, and Annalisa Fasolino
J. Phys.: Condens. Matter 24, 445009 (2012).
Joost A. van den Ende, Astrid S. de Wijn, and Annalisa Fasolino
J. Phys.: Condens. Matter 24, 445009 (2012).
We study the effects of temperature and sliding velocity on superlubricity in numerical simulations of the Frenkel–Kontorova model. We show that resonant excitations of the phonons in an incommensurate sliding body lead to an effective friction and to thermal equilibrium with energy distributed over the internal degrees of freedom. For finite temperature, the effective friction can be described well in terms of a viscous damping force, with a damping coefficient that emerges naturally from the microscopic dynamics. This damping coefficient is a non-monotonic function of the sliding velocity which peaks around resonant velocities and increases with temperature. At low velocities, it remains finite and nonzero, indicating the preservation of superlubricity in the zero-velocity limit. Finally, we propose experimental systems in which our results could be verified.
(In)commensurability, scaling, and multiplicity of friction in nanocrystals and application to gold nanocrystals on graphite
Astrid S. de Wijn
Phys. Rev. B 86, 085429 (2012).
Astrid S. de Wijn
Phys. Rev. B 86, 085429 (2012).
The scaling of friction with the contact size A and (in)commensurabilty of nanoscopic and mesoscopic crystals on a regular substrate are investigated analytically for triangular nanocrystals on hexagonal substrates. The crystals are assumed to be stiff, but not completely rigid. Commensurate and incommensurate configurations are identified systematically. It is shown that three distinct friction branches coexist, an incommensurate one that does not scale with the contact size (A^0) and two commensurate ones which scale differently (with A^(1/2) and A) and are associated with various combinations of commensurate and incommensurate lattice parameters and orientations. This coexistence is a direct consequence of the two-dimensional nature of the contact layer, and such multiplicity exists in all geometries consisting of regular lattices. To demonstrate this, the procedure is repeated for rectangular geometry. The scaling of irregularly shaped crystals is also considered, and again three branches are found (A^(1/4), A^(3/4), A). Based on the scaling properties, a quantity is defined which can be used to classify commensurability in infinite as well as finite contacts. Finally, the consequences for friction experiments on gold nanocrystals on graphite are discussed.
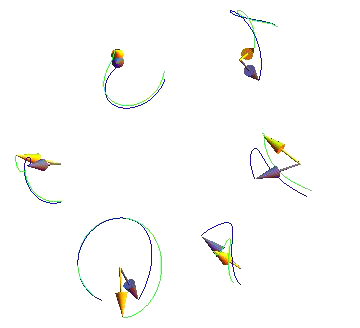
Largest Lyapunov Exponents for Lattices of Interacting Classical Spins
A. S. de Wijn, B. Hess, and B. V. Fine
Phys. Rev. Lett. 109, 034101 (2012).
A. S. de Wijn, B. Hess, and B. V. Fine
Phys. Rev. Lett. 109, 034101 (2012).
We investigate how generic the onset of chaos in interacting many-body classical systems is in the context of lattices of classical spins with nearest-neighbor anisotropic couplings. Seven large lattices in different spatial dimensions were considered. For each lattice, more than 2000 largest Lyapunov exponents for randomly sampled Hamiltonians were numerically computed. Our results strongly suggest the absence of integrable nearest-neighbor Hamiltonians for the infinite lattices except for the trivial Ising case. In the vicinity of the Ising case, the largest Lyapunov exponents exhibit a power-law growth, while further away they become rather weakly sensitive to the Hamiltonian anisotropy. We also provide an analytical derivation of these results.
Viscosity of liquid mixtures: The Vesovic-Wakeham method for chain molecules
Astrid S. de Wijn, Nicolas Riesco, George Jackson, J. P. Martin Trusler, and Velisa Vesovic
J. Chem. Phys. 136, 074514 (2012).
Astrid S. de Wijn, Nicolas Riesco, George Jackson, J. P. Martin Trusler, and Velisa Vesovic
J. Chem. Phys. 136, 074514 (2012).
New expressions for the viscosity of liquid mixtures, consisting of chain-like molecules, are derived by means of Enskog-type analysis. The molecules of the fluid are modelled as chains of equally sized, tangentially joined, and rigid spheres. It is assumed that the collision dynamics in such a fluid can be approximated by instantaneous collisions. We determine the molecular size parameters from the viscosity of each pure species and show how the different effective parameters can be evaluated by extending the Vesovic-Wakeham (VW) method. We propose and implement a number of thermodynamically consistent mixing rules, taking advantage of SAFT-type analysis, in order to develop the VW method for chain molecules. The predictions of the VW-chain model have been compared in the first instance with experimental viscosity data for octane-dodecane and methane-decane mixtures, thus, illustrating that the resulting VW–chain model is capable of accurately representing the viscosity of real liquid mixtures.
Spectral Narrowing in Coherent Rayleigh-Brillouin Scattering
A. Manteghi, N. J. Dam, A. S. Meijer, A. S. de Wijn, and W. van de Water
Phys. Rev. Lett. 107, 173903 (2011).
A. Manteghi, N. J. Dam, A. S. Meijer, A. S. de Wijn, and W. van de Water
Phys. Rev. Lett. 107, 173903 (2011).
Coherent Rayleigh-Brillouin scattering is a four-wave mixing technique that provides information on various physical properties of the scattering medium in the spectral domain. Being based on density gratings generated by dipole forces, the method requires two pump beams of sufficient spectral width to span the full response bandwidth of the scattering medium. We provide experimental data on the scattered spectrum as a function of the coherence between the two pump beams and derive the corresponding pump beam spectrum. We argue that all experiments on coherent Rayleigh-Brillouin scattering to date, have, in fact, been performed in the incoherent regime and show that orders of magnitude in scattering efficiency are to be expected when the experiments are performed with bandwidth-limited picosecond laser pulses.

Low friction and rotational dynamics of crystalline flakes in solid lubrication
Astrid S. de Wijn, Annalisa Fasolino, A. E. Filippov, and M. Urbakh
EPL 95, 66002 (2011).
Astrid S. de Wijn, Annalisa Fasolino, A. E. Filippov, and M. Urbakh
EPL 95, 66002 (2011).
Solids at incommensurate contact display low-friction, "superlubric", sliding. For graphene flakes on a graphite surface, superlubric sliding is only temporary due to rotation of the flakes from incommensurate to commensurate contact with the substrate. We examine this rotational channel of friction in a prototype geometry of meso- and macroscopic solid lubrication. By molecular-dynamics simulations and theoretical arguments we find that two surfaces lubricated by mobile, rotating graphene flakes exhibit stable superlubric sliding as for ideally incommensurate contacts also when they are covered by randomly oriented pinned graphene patches. For commensurate surfaces, we find a low-friction state at low temperature where incommensurate states are not destroyed by thermal fluctuations.
A radius of curvature approach to the Kolmogorov–Sinai entropy of dilute hard particles in equilibrium
Astrid S. de Wijn and Henk van Beijeren
J. Stat. Mech. P08012 (2011).
Astrid S. de Wijn and Henk van Beijeren
J. Stat. Mech. P08012 (2011).
We consider the Kolmogorov–Sinai entropy for dilute gases of N hard disks or spheres. This can be expanded in density as hₖₛ ∝ nN [ln(na^d) + B + O(na^d) + O(1/N)] , with a the diameter of the sphere or disk, n the density, and d the dimensionality of the system. We estimate the constant B by solving a linear differential equation for the approximate distribution of eigenvalues of the inverse radius of curvature tensor. We compare the resulting values of B both to previous estimates and to existing simulation results, finding very good agreement with the latter. Also, we compare the distribution of eigenvalues of the inverse radius of curvature tensor resulting from our calculations to new simulation results. For most of the spectrum the agreement between our calculations and the simulations again is very good.
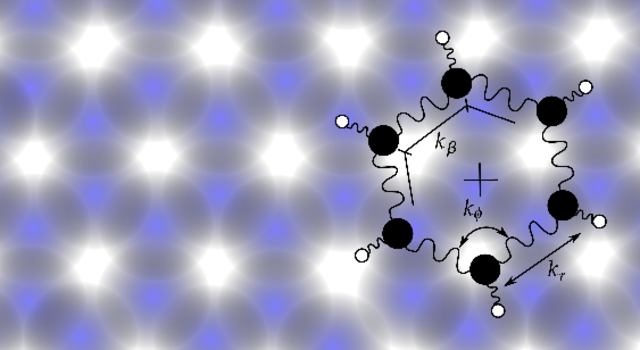
Internal degrees of freedom and transport of benzene on graphite
Astrid S. de Wijn
Phys. Rev. E 84, 011610 (2011).
Astrid S. de Wijn
Phys. Rev. E 84, 011610 (2011).
In this paper, the chaotic internal degrees of freedom of a benzene molecule adsorbed on a graphite substrate, their interplay with thermal noise, and their effects on the diffusion and drift are investigated analytically by making use of the presence of two different time scales as well as by molecular-dynamics simulations. The effects of thermal noise are investigated, and it is found that noise does not significantly alter the dynamics of the internal degrees of freedom yet does affect the friction and diffusion of the center of mass. Qualitative and quantitative theoretical predictions for the friction and diffusion of the molecule on the substrate are made and are compared to molecular-dynamics simulations. Contributions to the friction and diffusion from the finite heat bath as well as the slow dynamics of the center of mass are formally identified. It is shown that the torsion in benzene, which dominates the nonlinear coupling, significantly affects the friction of the molecule on the surface. The results compare favorably with recent results from He-neutron spin echo experiments on this system. Based on the analytical and numerical results, some suggestions are made for experimental conditions under which the effects of internal degrees of freedom might be observable.
Coherent and spontaneous Rayleigh-Brillouin scattering in atomic and molecular gases and gas mixtures
M. O. Vieitez, E. J. van Duijn, W. Ubachs, B. Witschas, A. Meijer, A. S. de Wijn, N. J. Dam, and W. van de Water
Phys. Rev. A 82, 043836 (2010).
M. O. Vieitez, E. J. van Duijn, W. Ubachs, B. Witschas, A. Meijer, A. S. de Wijn, N. J. Dam, and W. van de Water
Phys. Rev. A 82, 043836 (2010).
We study Rayleigh-Brillouin scattering in gases of N₂, O₂, and SF₆ molecules, Kr atoms, and He-Xe and He-CO₂ mixtures at pressures ranging from 1 to 3 bar and using two different experimental setups. In one setup, we measure spectra of light scattered by thermal density fluctuations (spontaneous Rayleigh-Brillouin scattering); in the second setup density waves are induced in the overlap region of two counterpropagating laser beams (coherent Rayleigh-Brillouin scattering). We compare measured spectra to the Tenti models and to a recent model for mixtures. We find new values of the bulk viscosity, which is a parameter in line-shape models that allows for internal degrees of freedom. Both experiments agree on the value of the bulk viscosity. Our results indicate a need for new line-shape models for mixtures of molecules with internal degrees of freedom.
Coherent Rayleigh–Brillouin scattering measurements of bulk viscosity of polar and nonpolar gases, and kinetic theory
A. S. Meijer, A. S. de Wijn, M. F. E. Peters, N. J. Dam, and W. van de Water
J. Chem. Phys. 133, 164315 (2010).
A. S. Meijer, A. S. de Wijn, M. F. E. Peters, N. J. Dam, and W. van de Water
J. Chem. Phys. 133, 164315 (2010).
We investigate coherent Rayleigh–Brillouin spectroscopy as an efficient process to measure the bulk viscosity of gases at gigahertz frequencies. Scattered spectral distributions are measured using a Fizeau spectrometer. We discuss the statistical error due to the fluctuating mode structure of the used pump laser. Experiments were done for both polar and nonpolar gases and the bulk viscosity was obtained from the spectra using the Tenti S6 model. Results are compared to simple classical kinetic models of molecules with internal degrees of freedom. At the extremely high (gigahertz) frequencies of our experiment, most internal vibrational modes remain frozen and the bulk viscosity is dominated by the rotational degrees of freedom. Our measurements show that the molecular dipole moments have unexpectedly little influence on the bulk viscosity at room temperature and moderate pressure.
Equivalence of kinetic-theory and random-matrix approaches to Lyapunov spectra of hard-sphere systems
Astrid S. de Wijn
Physica D: Nonlinear Phenomena 239, 1834–1841 (2010).
Astrid S. de Wijn
Physica D: Nonlinear Phenomena 239, 1834–1841 (2010).
In the study of chaotic behaviour of systems of many hard spheres, Lyapunov exponents of small absolute values exhibit interesting characteristics leading to speculations about connections to non-equilibrium statistical mechanics. Analytical approaches to these exponents so far can be divided into two groups, macroscopically oriented approaches, using kinetic theory or hydrodynamics, and more microscopically oriented random-matrix approaches in quasi-one-dimensional systems. In this paper, I present an approach using random matrices and weak-disorder expansion in an arbitrary number of dimensions. Correlations between subsequent collisions of a particle are taken into account. It is shown that the results are identical to those of a previous approach based on an extended Enskog equation. I conclude that each approach has its merits, and provides different insights into the approximations made, which include the Stoßzahlansatz, the continuum limit, and the long wavelength approximation. The comparison also gives insight into possible connections between Lyapunov exponents and fluctuations.
Stability of Low-Friction Surface Sliding of Nanocrystals with Rectangular Symmetry and Application to W on NaF(001)
Astrid S. de Wijn and Annalisa Fasolino
Tribology Letters 39, 91-99 (2010).
Astrid S. de Wijn and Annalisa Fasolino
Tribology Letters 39, 91-99 (2010).
We investigate the stability of low-friction sliding of nanocrystal with rectangular atomic arrangement on rectangular lattices, for which analytical results can be obtained. We find that several incommensurate periodic orbits exist and are stable against thermal fluctuations and other perturbations. As incommensurate orientations lead to low corrugation, and therefore low friction, such incommensurate periodic orbits are interesting for the study of nanotribology. The analytical results compare very well with simulations of W nanocrystals on NaF(001). The geometry and high typical corrugation of substrates with square lattices increase the robustness when compared to typical hexagonal lattices, such as graphite.
Stability of superlubric sliding on graphite
Astrid S. de Wijn, Claudio Fusco, and Annalisa Fasolino
Phys. Rev. E 81, 046105 (2010).
Astrid S. de Wijn, Claudio Fusco, and Annalisa Fasolino
Phys. Rev. E 81, 046105 (2010).
Recent atomic force microscope (AFM) experiments have shown that the low-friction sliding of incommensurate graphite flakes on graphite can be destroyed by torque-induced rotations. Here we theoretically investigate the stability of superlubric sliding against rotations of the flake. We find that the occurrence of superlubric motion critically depends on the physical parameters and on the experimental conditions: particular scan lines, thermal fluctuations, and high loading forces can destroy the stability of superlubric orbits. We find that the optimal conditions to achieve superlubric sliding are given by large flakes, low temperature, and low loads, as well as scanning velocities higher than those used in AFM experiments.
Relating chaos to deterministic diffusion of a molecule adsorbed on a surface
Astrid S. de Wijn and A. Fasolino
J. Phys.: Condens. Matter 21 264002 (2009).
Astrid S. de Wijn and A. Fasolino
J. Phys.: Condens. Matter 21 264002 (2009).
Chaotic internal degrees of freedom of a molecule can act as noise and affect the diffusion of the molecule on a substrate. A separation of timescales between the fast internal dynamics and the slow motion of the centre of mass on the substrate makes it possible to directly link chaos to diffusion. We discuss the conditions under which this is possible, and show that in simple atomistic models with pair-wise harmonic potentials, strong chaos can arise through the geometry. Using molecular dynamics simulations, we demonstrate that a realistic model of benzene is indeed chaotic, and that the internal chaos affects the diffusion on a graphite substrate.
A kinetic theory description of the viscosity of dense fluids consisting of chain molecules
Astrid S. de Wijn, Velisa Vesovic, George Jackson, and J. P. Martin Trusler
J. Chem. Phys. 128, 204901 (2008).
Astrid S. de Wijn, Velisa Vesovic, George Jackson, and J. P. Martin Trusler
J. Chem. Phys. 128, 204901 (2008).
An expression for the viscosity of a dense fluid is presented that includes the effect of molecular shape. The molecules of the fluid are approximated by chains of equal-sized, tangentially jointed, rigid spheres. It is assumed that the collision dynamics in such a fluid can be approximated by instantaneous collisions between two rigid spheres belonging to different chains. The approach is thus analogous to that of Enskog for a fluid consisting of rigid spheres. The description is developed in terms of two molecular parameters, the diameter σ of the spherical segment and the chain length (number of segments) m. It is demonstrated that an analysis of viscosity data of a particular pure fluid alone cannot be used to obtain independently effective values of both σ and m. Nevertheless, the chain lengths of n-alkanes are determined by assuming that the diameter of each rigid sphere making up the chain can be represented by the diameter of a methane molecule. The effective chain lengths of n-alkanes are found to increase linearly with the number C of carbon atoms present. The dependence can be approximated by a simple relationship m = 1+(C−1)/3. The same relationship was reported within the context of a statistical associating fluid theory equation of state treatment of the fluid, indicating that both the equilibrium thermodynamic properties and viscosity yield the same value for the chain lengths of n-alkanes.
Strong-field ionization in time-dependent density functional theory
A. S. de Wijn, M. Lein, and S. Kümmel
EPL 84 43001 (2008).
A. S. de Wijn, M. Lein, and S. Kümmel
EPL 84 43001 (2008).
Ionization in a strong laser field is a prime example of non-perturbative, correlated electron dynamics. Simulating such processes on a first-principles basis is a major theoretical challenge. We demonstrate that time-dependent density functional theory (TDDFT) on the basis of a newly developed functional for the correlation potential incorporates the relevant electron interaction effects. The central idea of our approach is to exploit information about exact ground-state correlation in approximate time-dependent calculations. The new functional provides an accurate description of the paradigm problem of the double ionization of the Helium atom, showing that TDDFT is a viable tool for strong-field calculations.
Vertical chaos and horizontal diffusion in the bouncing-ball billiard
Astrid S. de Wijn and Holger Kantz
Phys. Rev. E 75, 046214 (2007).
Astrid S. de Wijn and Holger Kantz
Phys. Rev. E 75, 046214 (2007).
The bouncing-ball billiard is a low-dimensional system with which transport properties of real physical systems can be studied theoretically. We study the bouncing-ball billiard with nonconvex scatterers and small slopes. We show that between the horizontal and vertical motion there is a separation of time scales, which is controlled by the slope of the billiard. We apply the theory of time-scale separation developed by Kantz et al. Physica D 187 200 (2004) If the vertical motion is chaotic, the horizontal motion is diffusive, but if the vertical motion is (quasi)periodic, there is no diffusion. We confirm the results with numerical simulations. Hence, the order-chaos transition in the vertical degrees of freedom translates into a localization-delocalization transition for the horizontal motion.
Numerical aspects of real-space approaches to strong-field electron dynamics
Astrid S. de Wijn, Stephan Kümmel, and Manfred Lein
J. Comp. Phys 226, 89-103 (2007).
Astrid S. de Wijn, Stephan Kümmel, and Manfred Lein
J. Comp. Phys 226, 89-103 (2007).
Numerical methods for calculating strong-field, nonperturbative electron dynamics are investigated. Two different quantum–mechanical approaches are discussed: the time-dependent Schrödinger equation and time-dependent density functional theory. We show that when solving the time-dependent Schrödinger equation, small errors in the initial ground-state wave function can be magnified considerably during propagation. A scheme is presented to efficiently obtain the ground state with high accuracy. We further demonstrate that the commonly-used absorbing boundary conditions can severely influence the results. The requirements on the boundary conditions are somewhat less stringent in effective single-particle approaches such as time-dependent density functional theory. We point out how results from accurate wave-function based calculations can be used to improve the density functional description of long-ranged, nonlinear electron dynamics. We present details of a method to reconstruct, numerically, the full, unapproximated, Kohn–Sham potential from the density and current of the exact system.
Lyapunov spectra of billiards with cylindrical scatterers: Comparison with many-particle systems
Astrid S. de Wijn
Phys. Rev. E 72, 026216 (2005).
Astrid S. de Wijn
Phys. Rev. E 72, 026216 (2005).
The dynamics of a system consisting of many spherical hard particles can be described as a single point particle moving in a high-dimensional space with fixed hypercylindrical scatterers with specific orientations and positions. In this paper, the similarities in the Lyapunov exponents are investigated between systems of many particles and high-dimensional billiards. The billiards contain cylindrical scatterers which have isotropically distributed orientations and homogeneously distributed positions. The dynamics of the isotropic billiards are calculated using a Monte Carlo simulation, and a reorthogonalization process is used to find the Lyapunov exponents. The results are compared to numerical results for systems of many hard particles as well as the analytical results for the high-dimensional Lorentz gas. The smallest three-quarters of the positive exponents behave more like the exponents of hard-disk systems than the exponents of the Lorentz gas. This similarity shows that the hard-disk systems may be approximated by a spatially homogeneous and isotropic system of scatterers for a calculation of the smaller Lyapunov exponents, apart from the exponent associated with localization. The method of the partial stretching factor is used to calculate these exponents analytically, with results that compare well with simulation results of hard disks and hard spheres.
Kolmogorov-Sinai entropy for dilute systems of hard particles in equilibrium
Astrid S. de Wijn
Phys. Rev. E 71, 046211 (2005).
Astrid S. de Wijn
Phys. Rev. E 71, 046211 (2005).
In an equilibrium system, the Kolmogorov-Sinai entropy hₖₛ equals the sum of the positive Lyapunov exponents, the exponential rates of divergence of infinitesimal perturbations. Kinetic theory may be used to calculate the Kolmogorov-Sinai entropy for dilute gases of many hard disks or spheres in equilibrium at low number density n. The density expansion of hₖₛ is Nν̅A[lnn+B+O(n)], where ν̅ is the single-particle collision frequency. Previous calculations of A were successful. Calculations of B, however, were unsatisfactory. In this paper, I show how the probability distribution of the stretching factor can be determined from a nonlinear differential equation by an iterative method. From this the Kolmogorov-Sinai entropy follows as the average of the logarithm of the stretching factor per unit time. I calculate approximate values of B and compare these to results from existing simulations. The agreement is good.

Chaos in systems with many degrees of freedom
Astrid S. de Wijn
PhD thesis, Utrecht University (2004), ISBN 90-393-3868-X.
Astrid S. de Wijn
PhD thesis, Utrecht University (2004), ISBN 90-393-3868-X.
In this thesis I discuss some of the chaotic properties specific to systems of many particles and other systems with many degrees of freedom. A dynamical system is called chaotic if a typical infinite perturbation of initial conditions grows exponentially with time. The chaoticity of a system is characterised by the Lyapunov exponents, which indicate the possible rates at which an infinitesimal perturbation of initial conditions may grow or decrease. A system has as many Lyapunov exponents as it's phase space has dimensions, and so a system with many degrees of freedom has many Lyapunov exponents. A system is chaotic if it has at least one positive exponent. The sum of the positive Lyapunov exponents equals the maximal rate of information increase of the system and is referred to as the Kolmogorov-Sinai entropy. The dynamical properties of a system are thought to have bearing on the non-equilibrium behaviour. This thesis contains calculations of Lyapunov exponents of three different systems; namely, systems consisting of many, freely moving hard disks and hard spheres are discussed, as well as the Lorentz gas, which is a system consisting of fixed spherical scatterers with a point particle moving between and colliding elastically with them, and a similar system in which the scatterers are cylindrical. Chapter 3 contains calculations of the smallest positive and negative exponents of hard disks. These are known from simulations to have interesting behaviour, if the system is large enough. They are explained as belonging to Goldstone modes associated with the symmetries of the tangent space, and the Lyapunov exponents are calculated. This allows their calculation by formulating a generalised Boltzmann equation and solving this perturbatively. There is a slight discrepancy between the predicted values and simulation results, which may be attributed to the neglect of ring-collision terms in the Boltzmann equation. In chapter 4 the Kolmogorov-Sinai entropy is calculated for the same system. It is known to be proportional to the collision frequency multiplied by a factor that equals the logarithm of the density plus a constant. Previous calculations of this constant have produced unsatisfactory results. I discuss the cause of this and how to calculate the constant correctly. A system of freely moving hard particles can be described as a point particle in a high-dimensional space, colliding with cylindrical scatterers with very specific positions and orientations. Because this is very similar to the Lorentz gas, where the scatterers are hyperspheres, in chapter 5 I examine the Lyapunov exponents of the Lorentz gas in an arbitrary number of dimensions. The full spectrum is calculated analytically and the similarities and differences between the high-dimensional Lorentz gas and systems of hard disks or spheres are discussed. In chapter 6 I describe numerical calculations of the Lyapunov exponents of systems consisting of isotropically oriented, homogeneously distributed, cylindrical scatterers. Because of the similarity in the shape of the scatterers, the Lyapunov spectrum of this system is much more similar to the spectrum of hard disks than the spectrum of the Lorentz gas. Using the techniques developed in chapters 4 and 5, I estimate the smallest positive and negative Lyapunov exponents of hard disks and hard spheres which are not associated with Goldstone modes.
Lyapunov spectrum of the many-dimensional dilute random Lorentz gas
Astrid S. de Wijn and Henk van Beijeren
Phys. Rev. E 70, 036209 (2004).
Astrid S. de Wijn and Henk van Beijeren
Phys. Rev. E 70, 036209 (2004).
For a better understanding of the chaotic behavior of systems of many moving particles, it is useful to look at other systems with many degrees of freedom. An interesting example is the high-dimensional Lorentz gas, which, just like a system of moving hard spheres, may be interpreted as a dynamical system consisting of a point particle in a high-dimensional phase space, moving among fixed scatterers. In this paper, we calculate the full spectrum of Lyapunov exponents for the dilute random Lorentz gas in an arbitrary number of dimensions. We find that the spectrum becomes flatter with increasing dimensionality. Furthermore, for fixed collision frequency the separation between the largest Lyapunov exponent and the second largest one increases logarithmically with dimensionality, whereas the separations between Lyapunov exponents of given indices not involving the largest one go to fixed limits.
Goldstone modes in Lyapunov spectra of hard sphere systems
Astrid S. de Wijn and Henk van Beijeren
Phys. Rev. E 70, 016207 (2004).
Astrid S. de Wijn and Henk van Beijeren
Phys. Rev. E 70, 016207 (2004).
In the study of chaotic behavior, Lyapunov exponents play an important part. In this paper, we demonstrate how the Lyapunov exponents close to zero of a system of many hard spheres can be described as Goldstone modes, by using a Boltzmann type of approach. At low densities, the correct form is found for the wave number dependence of the exponents as well as for the corresponding eigenvectors in tangent space. The predicted values for the Lyapunov exponents belonging to the transverse mode are within a few percent of the values found in recent simulations, the propagation velocity for the longitudinal mode is within 1%, but the value for the Lyapunov exponent belonging to the longitudinal mode deviates from the simulations by 30%. For higher densities, the predicted values deviate more from the values calculated in the simulations. These deviations may be due to contributions from ring collisions and similar terms, which, even at low densities, can contribute to the leading order.